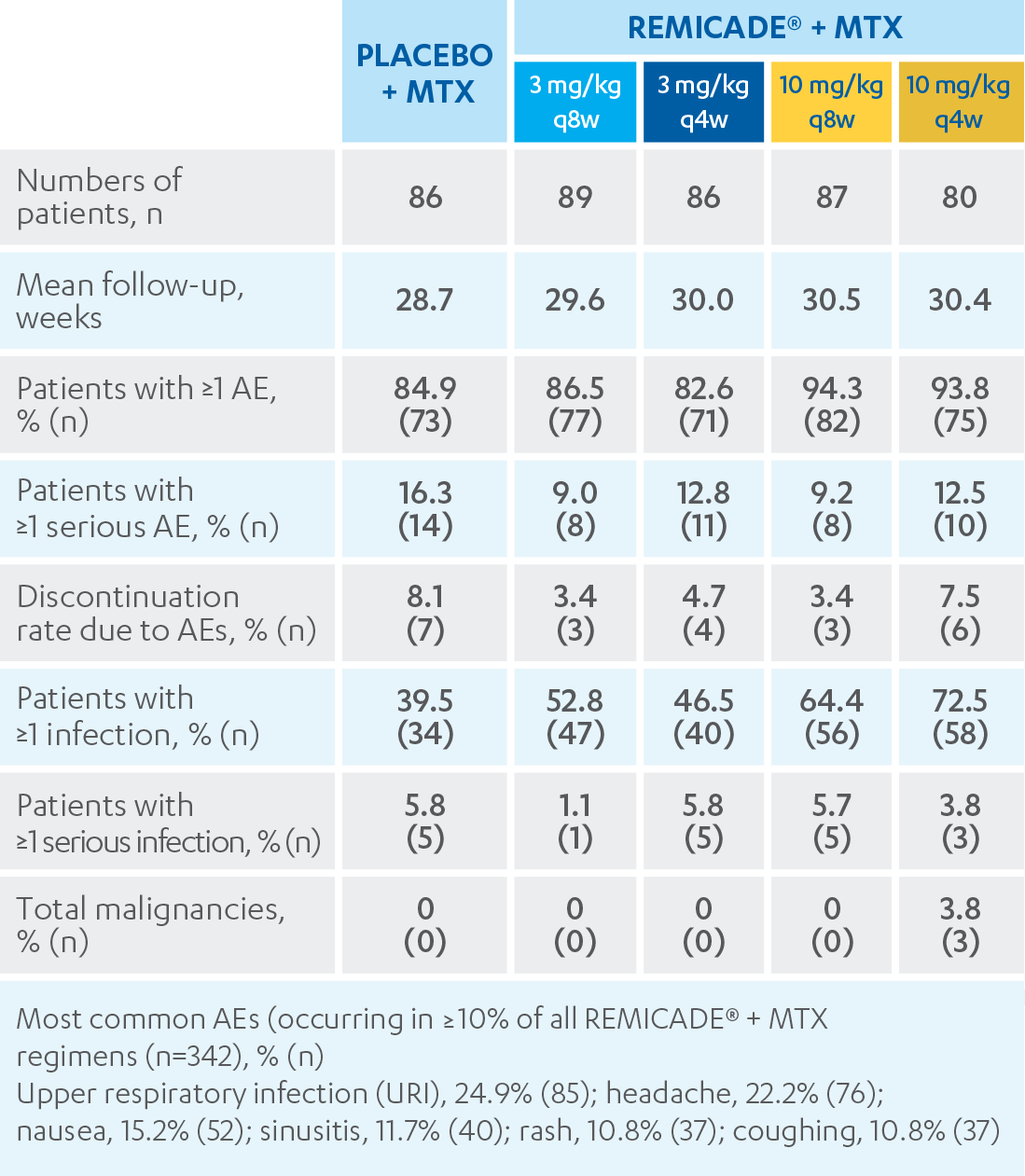



A COMPARATIVE AND PRAGMATIC STUDY
OF SIMPONI ARIA® (golimumab) vs REMICADE® (infliximab) IN RA
LIMITATIONS
- As with other open-label, nonrandomized RWE, there is the potential for selection, channeling, and other forms of bias, which may confound these results1
- Because the study was not randomized, differences in baseline (BL) characteristics between treatment groups were statistically addressed by utilizing propensity weighting methodology1
- Results are not intended for direct comparison with Phase 3 clinical trials because the real-world studies were observational trials. There have been no head-to-head randomized clinical trials comparing the safety and efficacy of SIMPONI ARIA® vs REMICADE®
STUDY METHODOLOGY
- These are data from a planned interim analysis of an ongoing Phase 4 study; the study will continue and will capture data through the anticipated 3-year follow-up1
- Hierarchical procedures were specified for the primary and major secondary endpoints at interim and final analyses to control α level at 0.051
- The major secondary endpoints (Clinical Disease Activity Index [CDAI] change from BL at months 6 and 12 in the bio-naÏve set) test the noninferiority of SIMPONI ARIA® compared to REMICADE® with pre-identified rules for missing data1

PRIMARY AND MAJOR SECONDARY ENDPOINTS WERE MET AT THE FORMAL INTERIM ANALYSIS
FORMAL INTERIM ANALYSIS WAS CONDUCTED AFTER APPROXIMATELY 66% OF ENROLLED SUBJECTS HAD COMPLETED THE 52-WEEK VISIT OR DISCONTINUED1,2

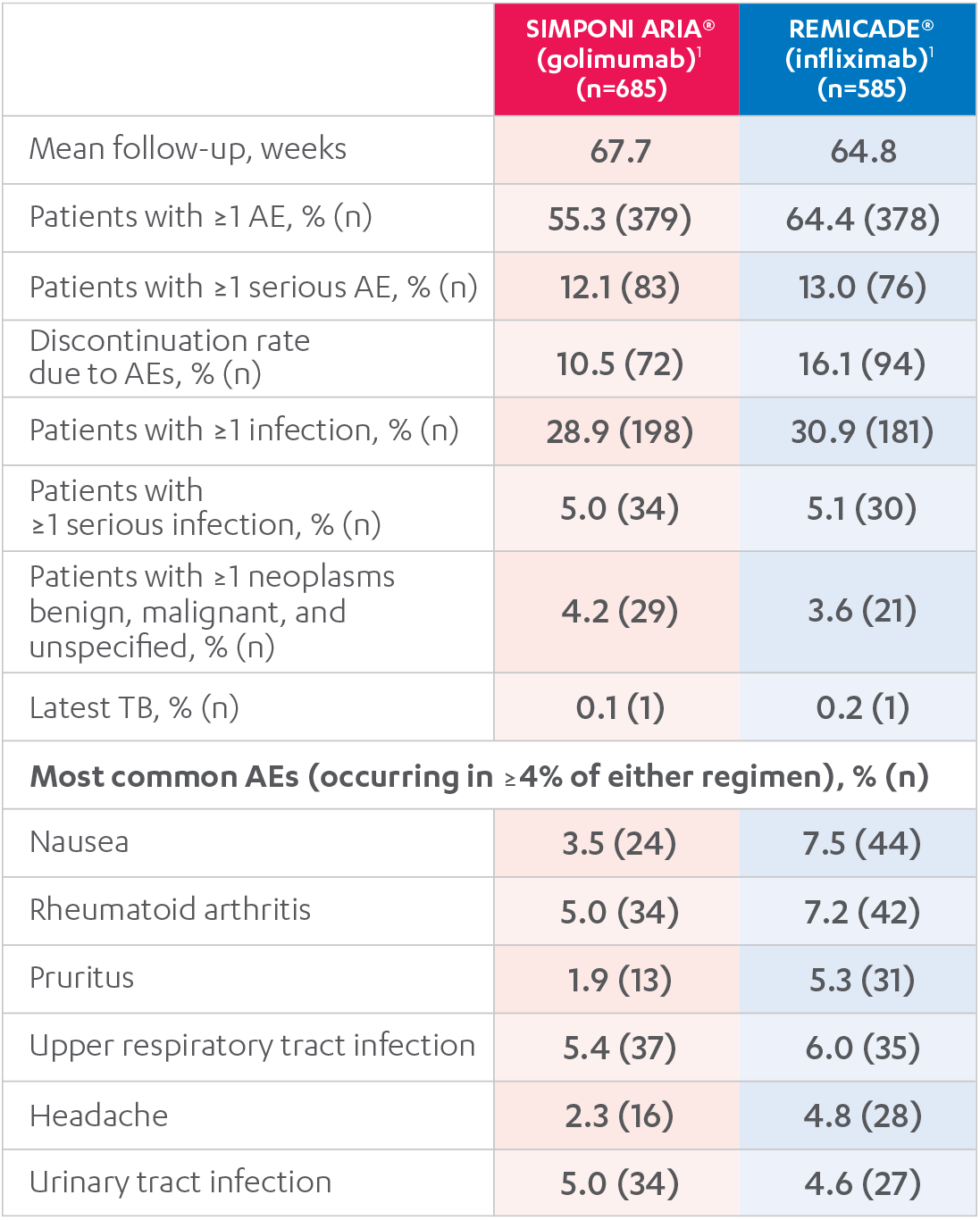
Results are not intended for direct comparison with Phase 3 clinical trials because the real-world studies were observational trials. There have been no head-to-head randomized clinical trials comparing the safety and efficacy of SIMPONI ARIA® vs REMICADE®.


Results are not intended for direct comparison with Phase 3 clinical trials because the real-world studies were observational trials. There have been no head-to-head randomized clinical trials comparing the safety and efficacy of SIMPONI ARIA® vs REMICADE®.
STUDY UPDATE: 52-WEEK DATA1
AWARE is an ongoing study beyond the formal interim analysis in which all endpoints were met.*
- All subsequent data reported from the 52-week data set are descriptive
- 52-week data include all patients with at least 52 weeks data or discontinued prior to Week 52
- 52-week data provide an update on:
- Infusion reactions
- Dose escalation
- Discontinuations
- Effectiveness
- Safety
- Endpoints were met at the formal interim analysis, therefore no further comparative statistical analyses were needed.
CI=confidence interval.
*Propensity score adjustments were completed for the primary and major secondary endpoints.
†Based on a noninferiority margin of 6.
‡Imputations for change from baseline CDAI values employed treatment failure rules and LOCF for missing data.
§95% CI for primary endpoint; 96.42% CI provided for secondary endpoints based on alpha spending function.
WHAT WERE THE BASELINE DEMOGRAPHICS
IN THE AWARE STUDY?
BASELINE DEMOGRAPHICS1,2
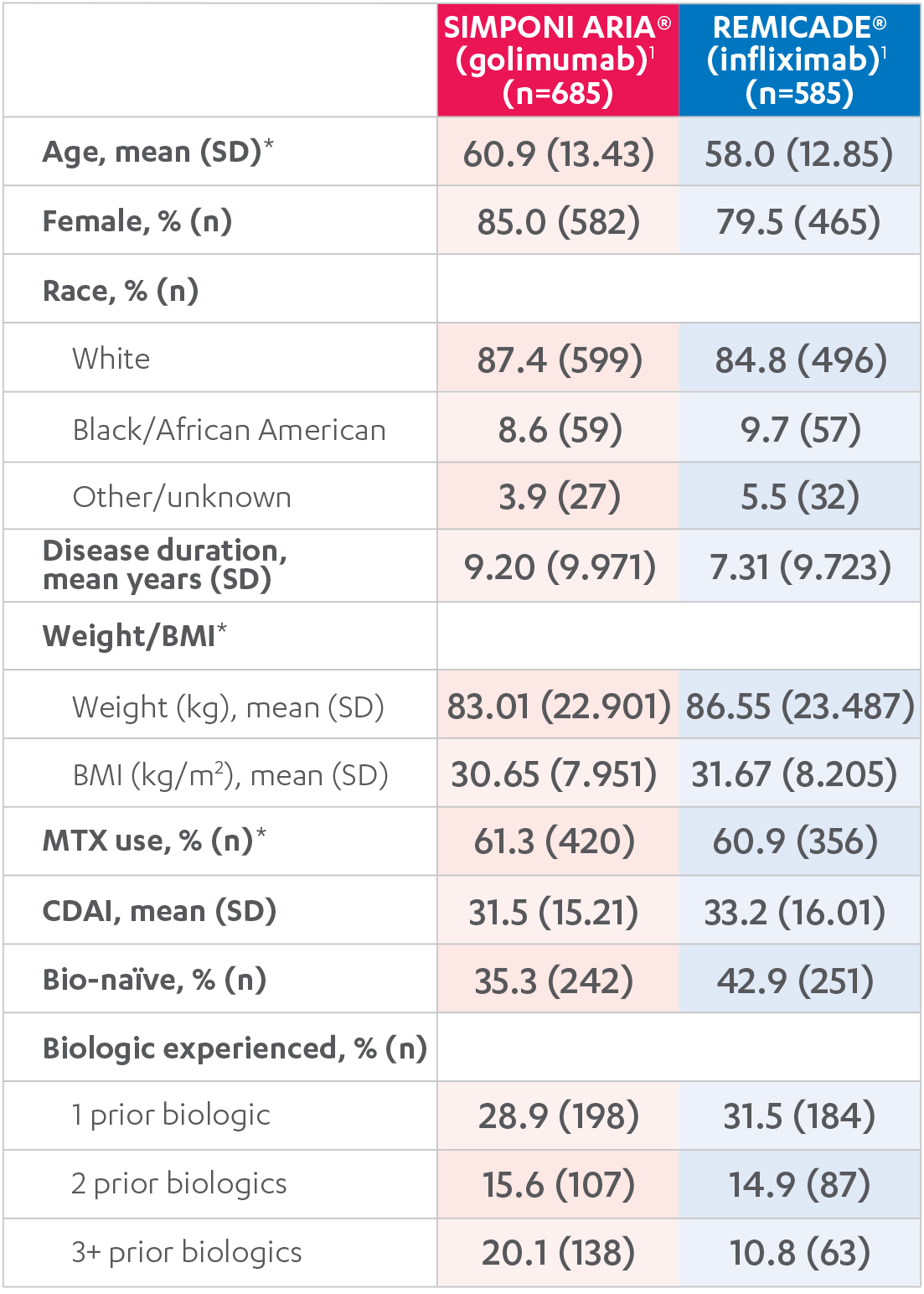
BMI=body mass index; SD=standard deviation.
*Baseline weight/BMI/CDAI were not available for all participants.
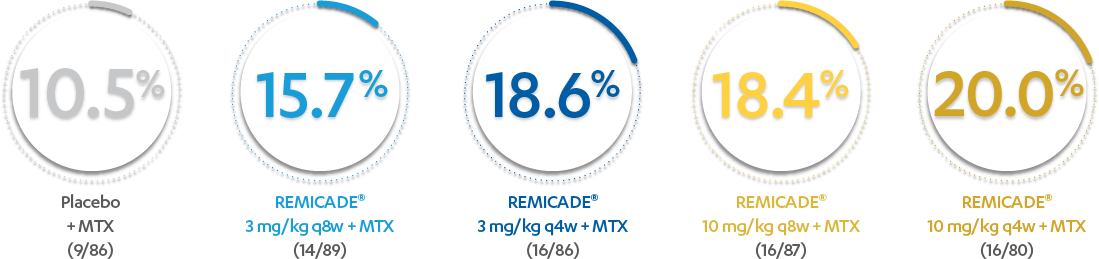
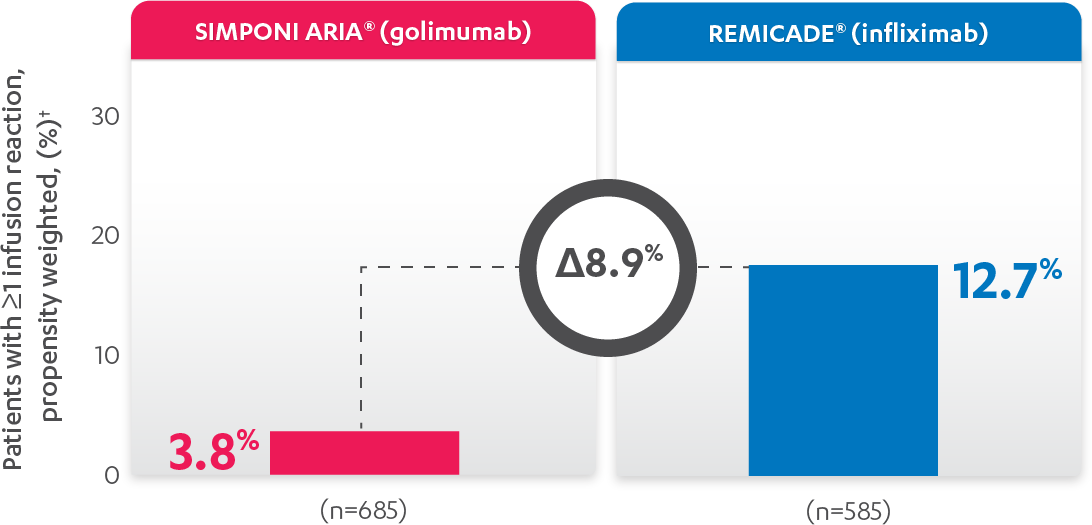
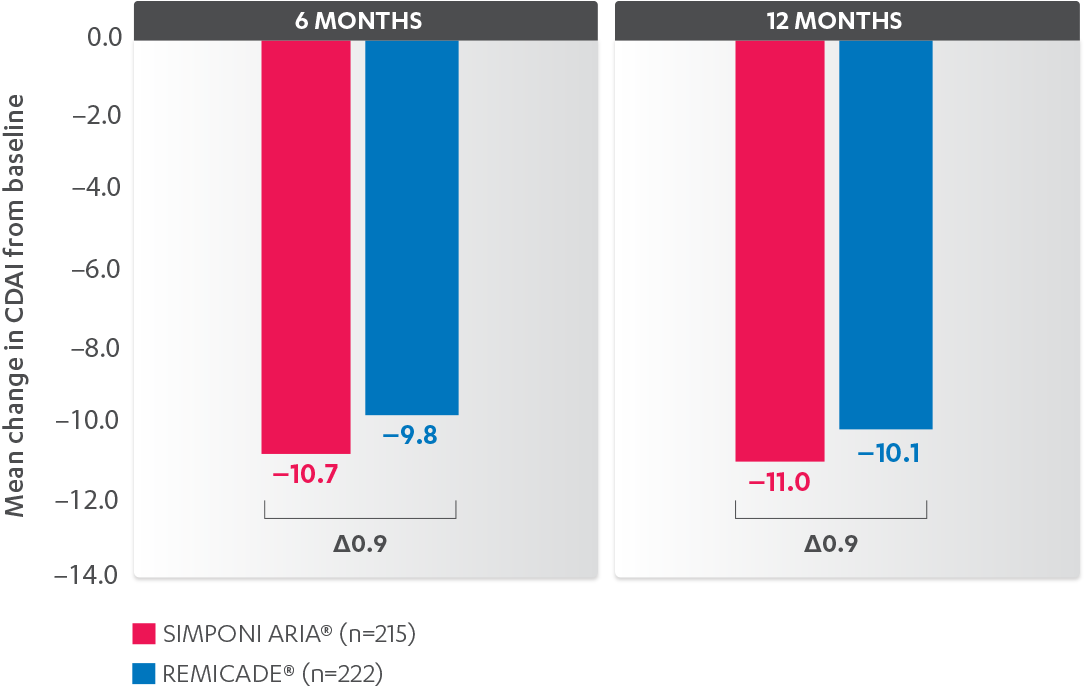
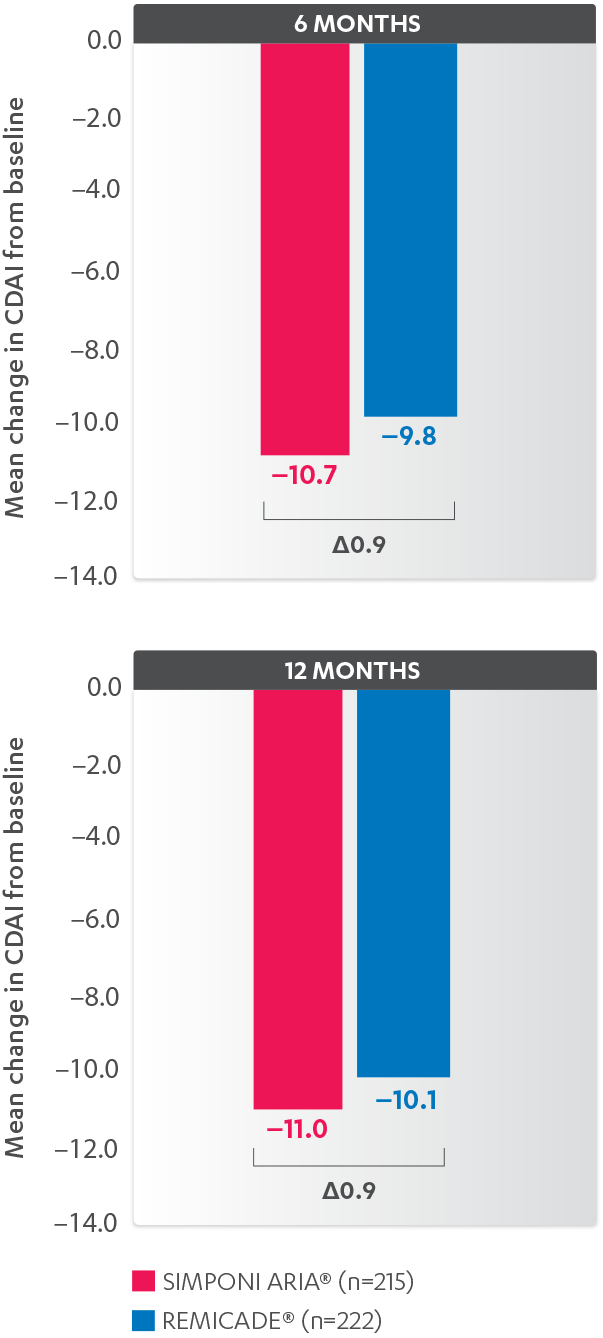
INFUSION REACTIONS AND CHANGE FROM BASELINE IN CDAI
IN RA, 52 WEEKS
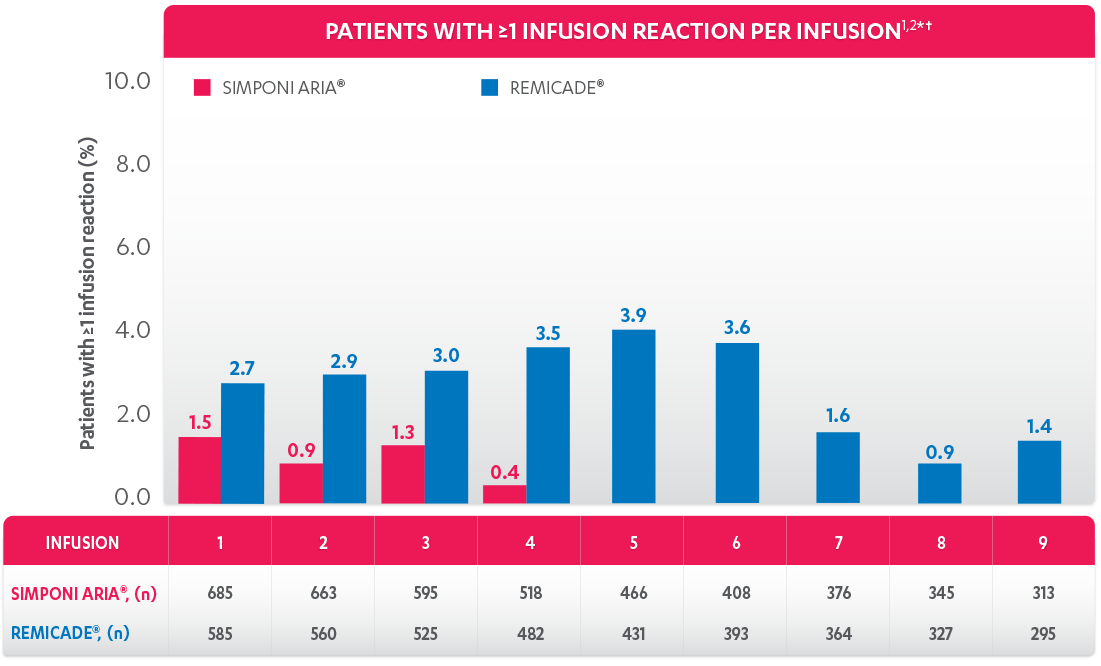
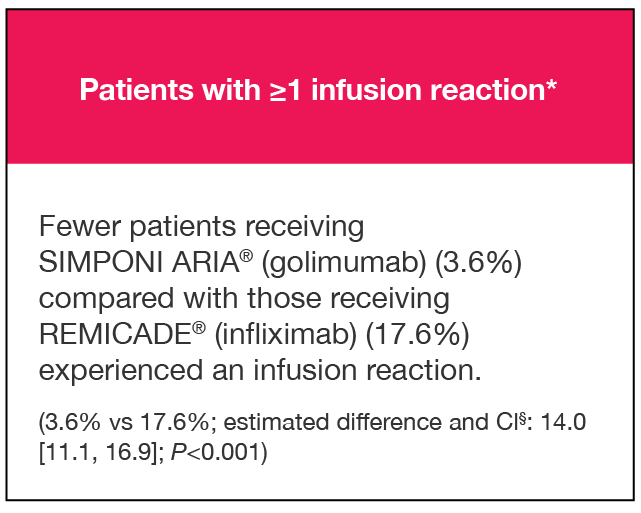
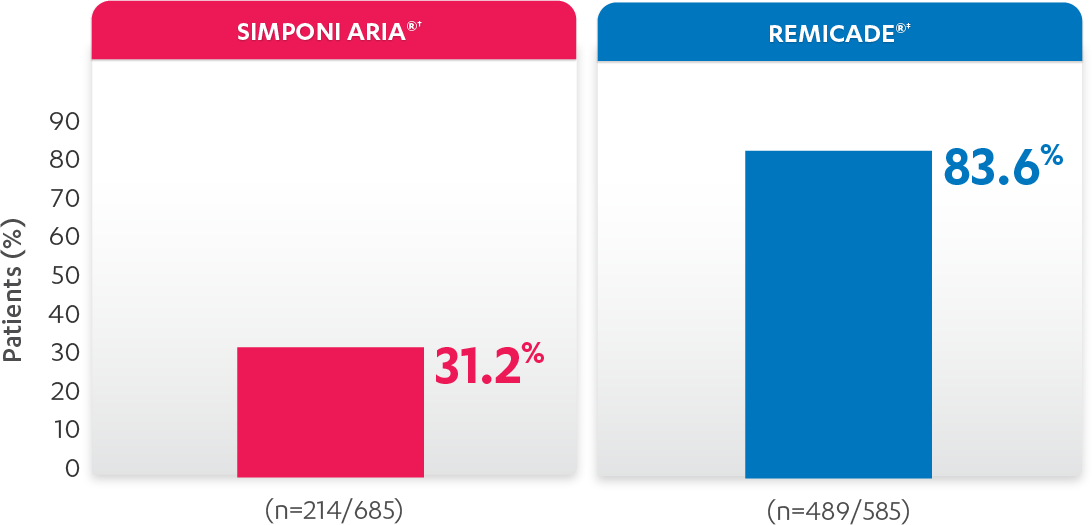
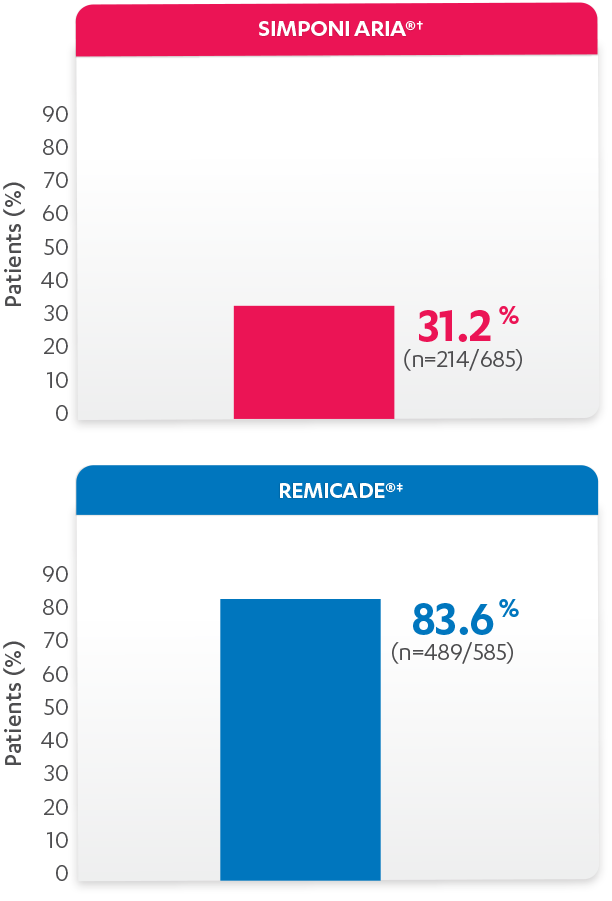
PERCENTAGE OF PATIENTS WITH ≥1 INFUSION REACTION:
PATIENTS WITH ≥1 INFUSION REACTION (%)1,2*†
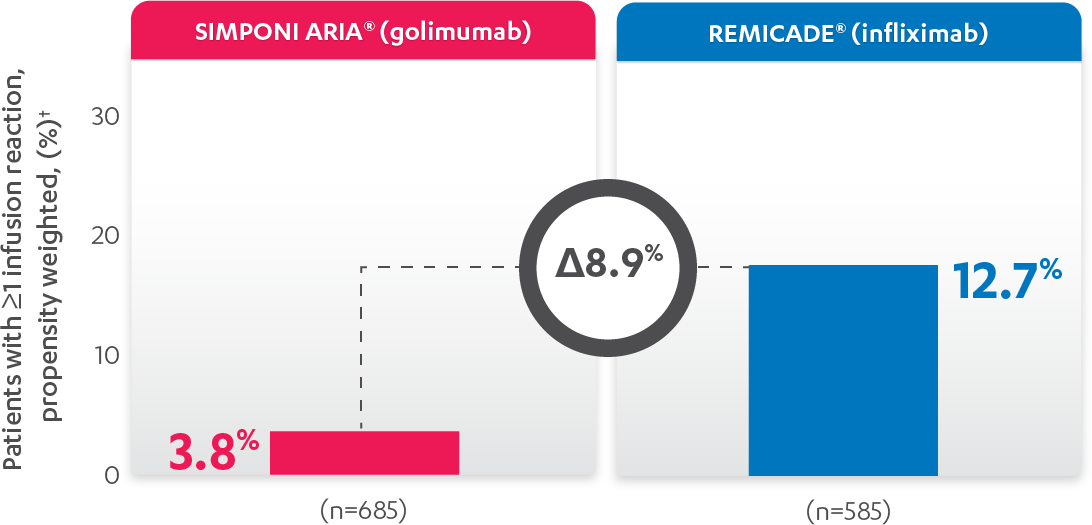
Results are not intended for direct comparison with Phase 3 clinical trials because the real-world studies were observational trials. There have been no head-to-head randomized clinical trials comparing the safety and efficacy of SIMPONI ARIA® vs REMICADE®.
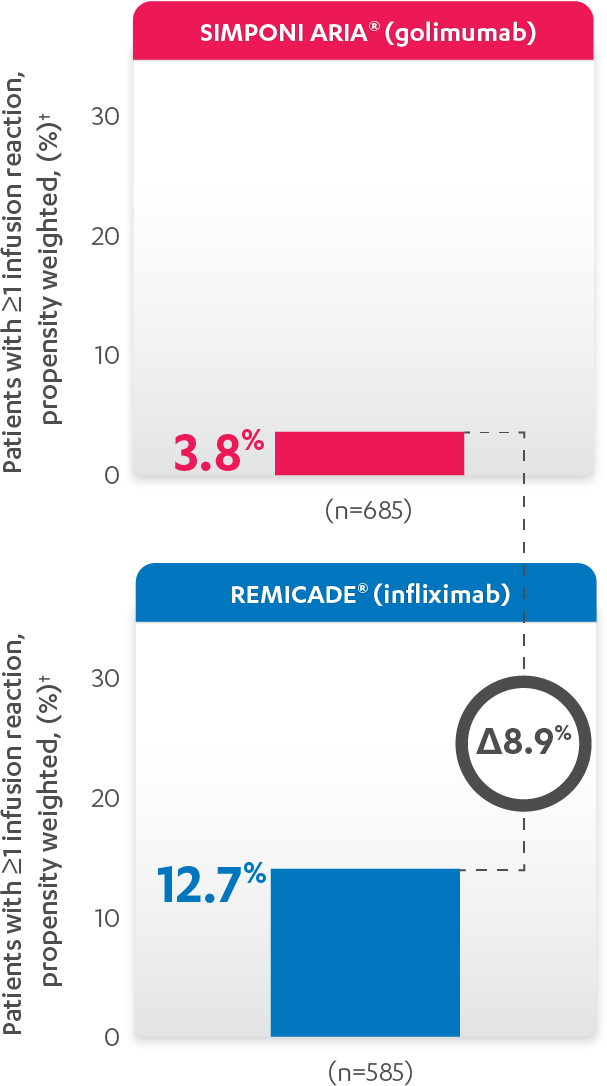
Results are not intended for direct comparison with Phase 3 clinical trials because the real-world studies were observational trials. There have been no head-to-head randomized clinical trials comparing the safety and efficacy of SIMPONI ARIA® vs REMICADE®.
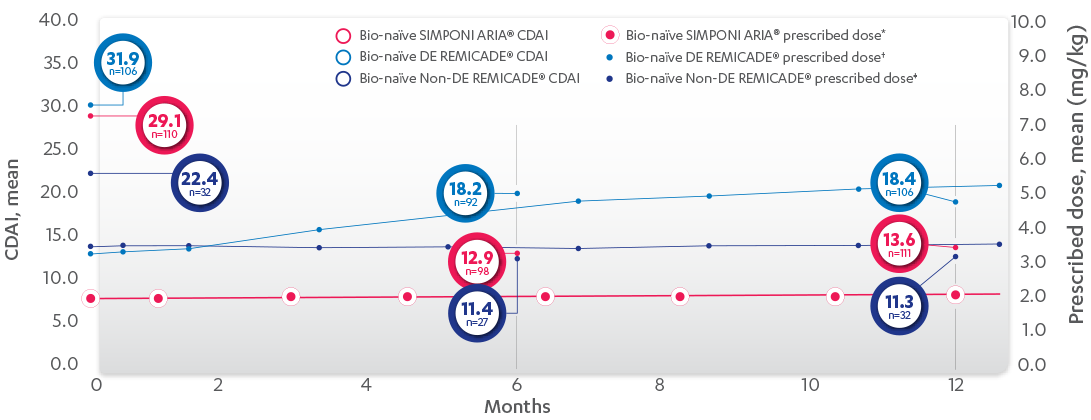
CDAI CHANGE FROM BASELINE AT 6 AND 12 MONTHS, BIO-NAÏVE1,2*†‡
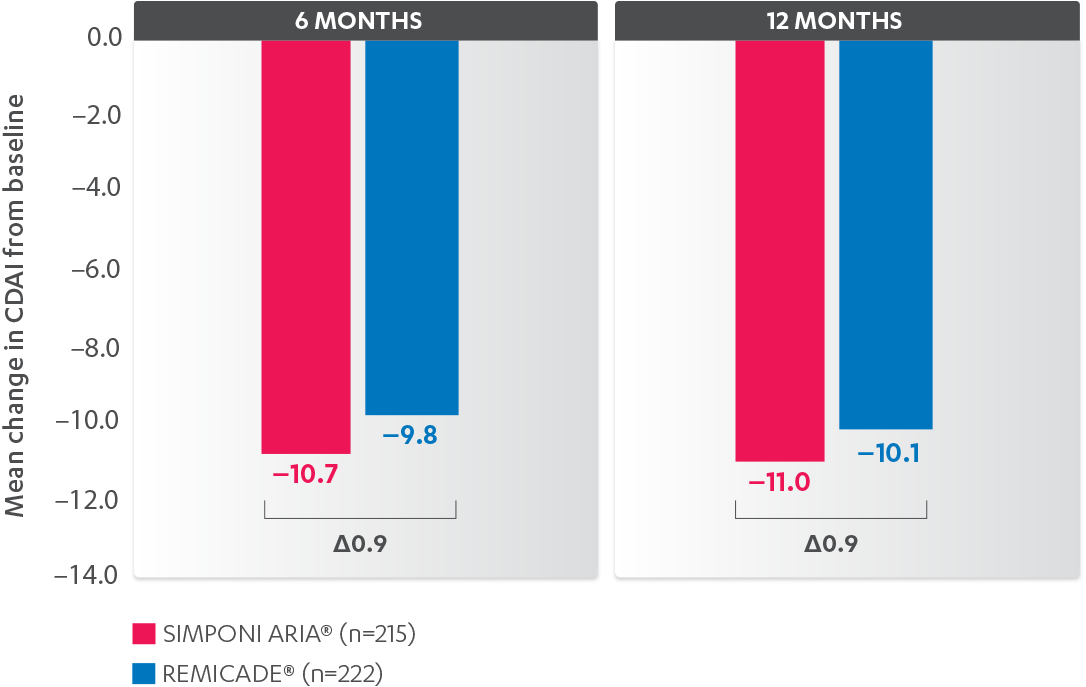
-
Results are not intended for direct comparison with Phase 3 clinical trials because the real-world studies were observational trials. There have been no head-to-head randomized clinical trials comparing the safety and efficacy of SIMPONI ARIA® vs REMICADE®.
*Propensity score adjustments were completed at 52 weeks for the endpoints, infusion reactions, and CDAI change from baseline at 6 and 12 months.
†An infusion reaction is any AE that occurs during an infusion, or within 1 hour after the infusion of either SIMPONI ARIA® or REMICADE®.
‡Imputations for change from baseline CDAI values employed treatment failure rules and LOCF for missing data.
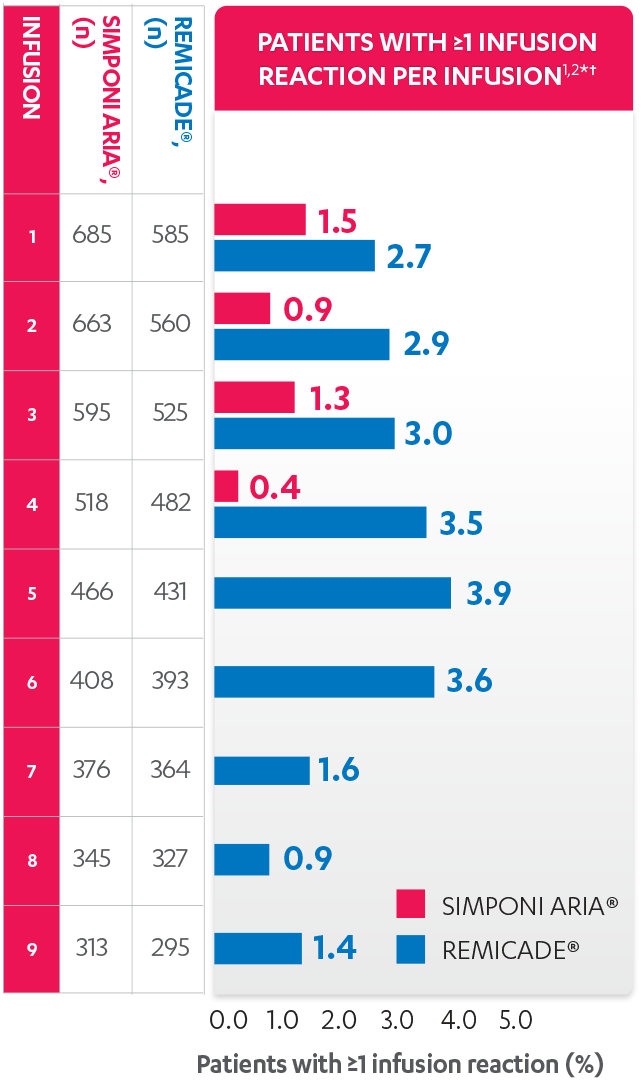
PATIENTS WITH AN INFUSION REACTION, PER INFUSION (%), 52 WEEKS1

-
Results are not intended for direct comparison with Phase 3 clinical trials because the real-world studies were observational trials. There have been no head-to-head randomized clinical trials comparing the safety and efficacy of SIMPONI ARIA® vs REMICADE®.
Patients receiving SIMPONI ARIA® had infusion reactions through the 4th infusion. Patients receiving REMICADE® had infusion reactions through the 9th infusion.
*An infusion reaction is any AE that occurs during an infusion, or within 1 hour after the infusion, of either SIMPONI ARIA® or REMICADE®.
†All patients that received at least 1 infusion of SIMPONI ARIA® or REMICADE® and have ≥52 weeks of treatment or discontinued prior to Week 52.
USE OF ≥1 PRE-INFUSION MEDICATION
IN RA THROUGH 52 WEEKS1,2*
-
Results are not intended for direct comparison with Phase 3 clinical trials because the real-world studies were observational trials. There have been no head-to-head randomized clinical trials comparing the safety and efficacy of SIMPONI ARIA® vs REMICADE®.
Study did not mandate the use of pre-infusion medication (pre-infusion medication usage occurred per usual clinical practice).
*Used at any time during the study.
†Pre-infusion medications are not part of the SIMPONI ARIA® Prescribing Information.
‡According to the REMICADE® Prescribing Information, premedication could include antihistamine (anti-H1 +/- anti-H2), acetaminophen, and/or corticosteroids.
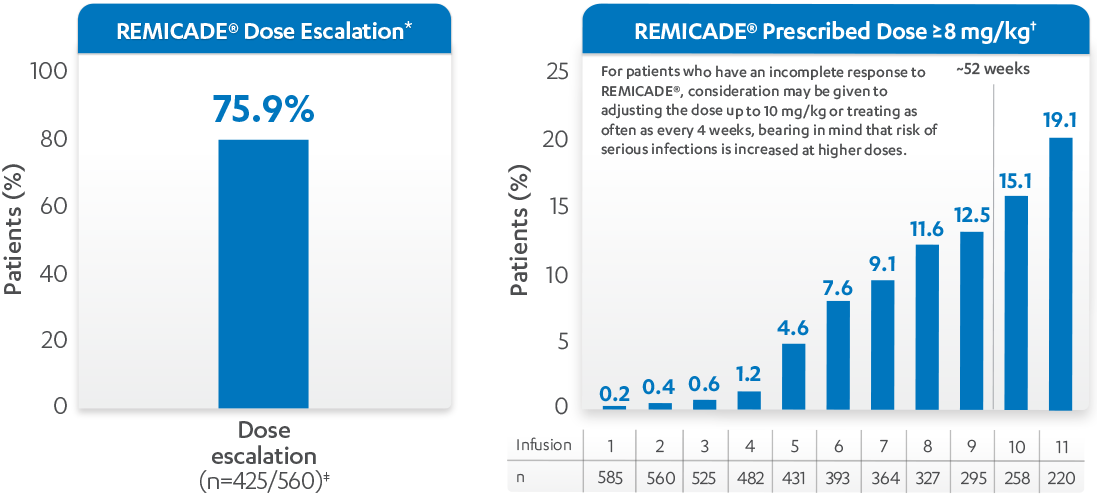
REMICADE® (infliximab) DOSE ESCALATION, 52 WEEKS
DOSE ESCALATION, 52 WEEKS
REMICADE®1*†
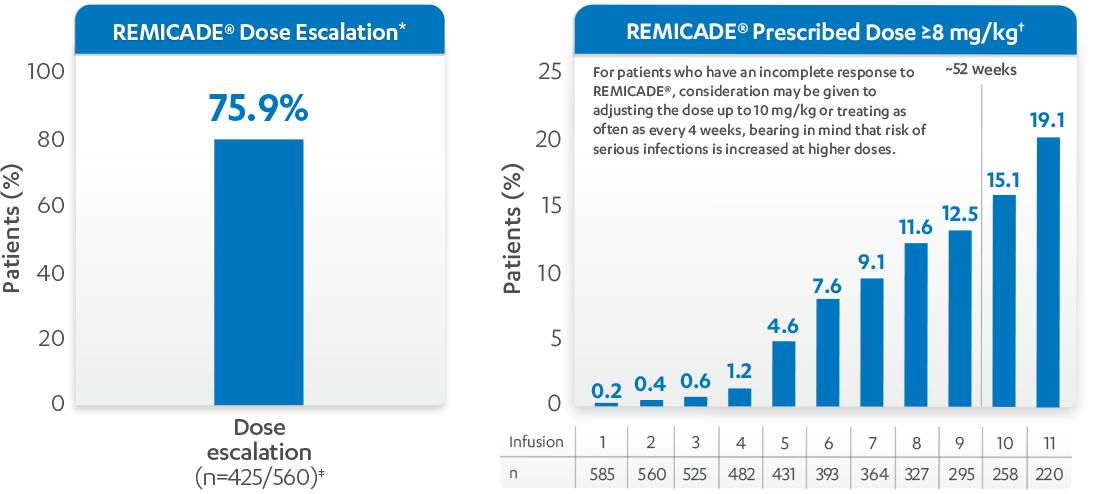

-
Results are not intended for direct comparison with Phase 3 clinical trials because the real-world studies were observational trials. There have been no head-to-head randomized clinical trials comparing the safety and efficacy of SIMPONI ARIA® vs REMICADE®.
*Dose escalation=percent of patients with at least 1 post-baseline normalized dose > baseline dose. Normalized dose was calculated for each patient by infusion visit. At baseline, normalized dose was the prescribed dose. After baseline, normalized dose was adjusted by the actual time interval from the previous visit: Normalized dose=prescribed dose x (scheduled time interval [week])/(actual time interval [week]) where the scheduled time interval is 2 (infusion visit 2), 4 (infusion visit 3), then every 8 weeks (from infusion visit 4 on), actual time interval is (infusion visit date–previous infusion visit date)/7. Summary statistics of the normalized dose were generated by treatment group and infusion visit.
†8 mg/kg is based on dose alone and not adjusting for dose interval.
‡Dosing and dosing interval data were not available for all patients.
REMICADE® DOSING:
REMICADE® is administered by IV infusion over a period of not less than 2 hours. The recommended dose of REMICADE® is 3 mg/kg given as an induction regimen at 0, 2, and 6 weeks, followed by a maintenance regimen of 3 mg/kg every 8 weeks thereafter for the treatment of moderately to severely active RA. REMICADE® should be given in combination with MTX.2
RELATIONSHIP BETWEEN MEAN PRESCRIBED DOSE AND CDAI, 52 WEEKS
FOR SIMPONI ARIA® (golimumab) AND REMICADE® (infliximab)
RELATIONSHIP BETWEEN MEAN PRESCRIBED DOSE AND CDAI IN BIO-NAÏVE PATIENTS, 52 WEEKS
PROPENSITY-ADJUSTED AND IMPUTED DATA*
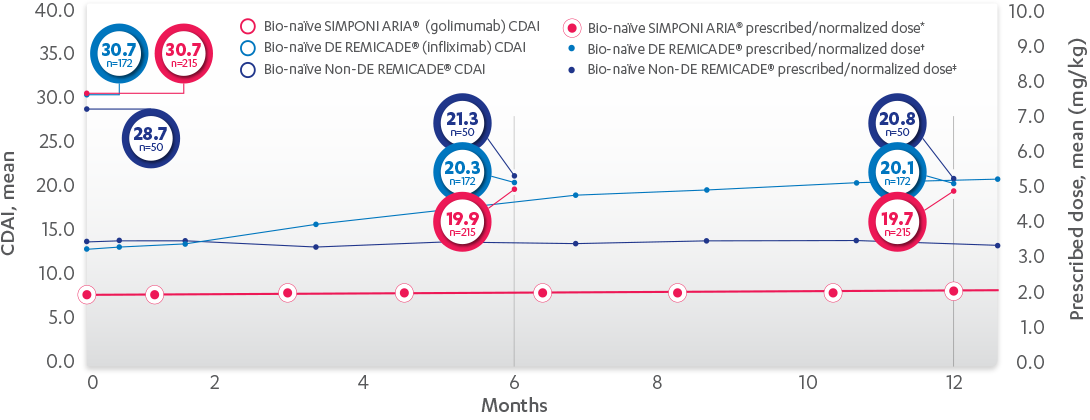
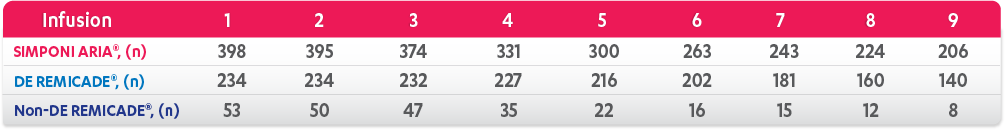
Patients Per Infusion
Results are not intended for direct comparison with Phase 3 clinical trials because the real-world studies were observational trials. There have been no head-to-head randomized clinical trials comparing the safety and efficacy of SIMPONI ARIA® vs REMICADE®.
SIMPONI ARIA® mean change† from baseline at 6 months=–10.8 and 12 months=–11.1. DE=at least 1 post-baseline normalized dose > baseline dose. REMICADE® DE IPTW imputed mean change from baseline at 6 months=–10.4 and 12 months=–10.7. REMICADE® non-DE imputed mean change from baseline at 6 months=–7.4 and 12 months=–7.9.
DE=dose escalation; IPTW=inverse probability of treatment weighting.
*LOCF with treatment failure rules.
†Means with weight on IPTW propensity score.
‡Patients receiving SIMPONI ARIA® at Months 0, 6, and 12 were on infusions 1, 4, and 7 respectively.
§Patients receiving DE REMICADE® at Month 0, 6, and 12 were on infusions 1, 5, and 8 respectively.
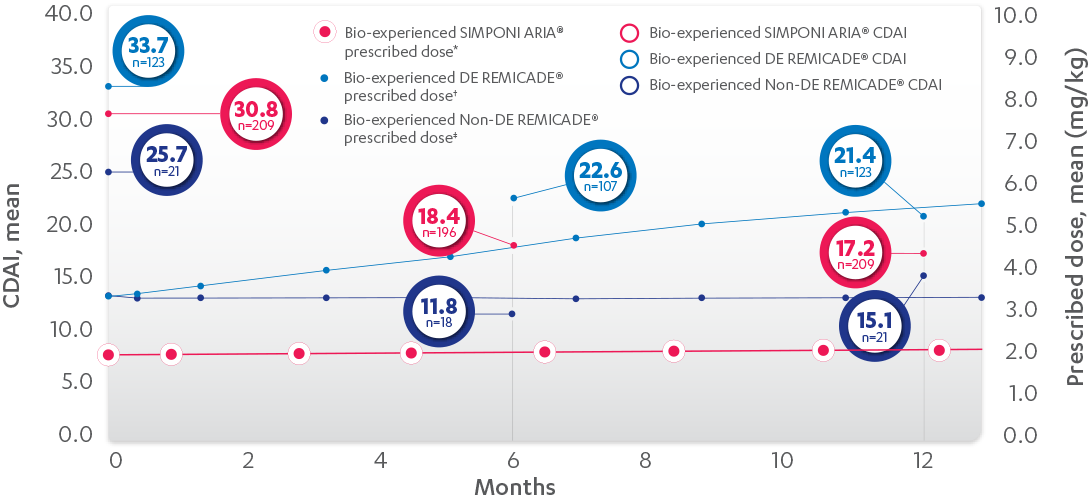
RELATIONSHIP BETWEEN MEAN PRESCRIBED DOSE AND CDAI IN BIO-EXPERIENCED PATIENTS, 52 WEEKS1
PROPENSITY-ADJUSTED AND IMPUTED DATA
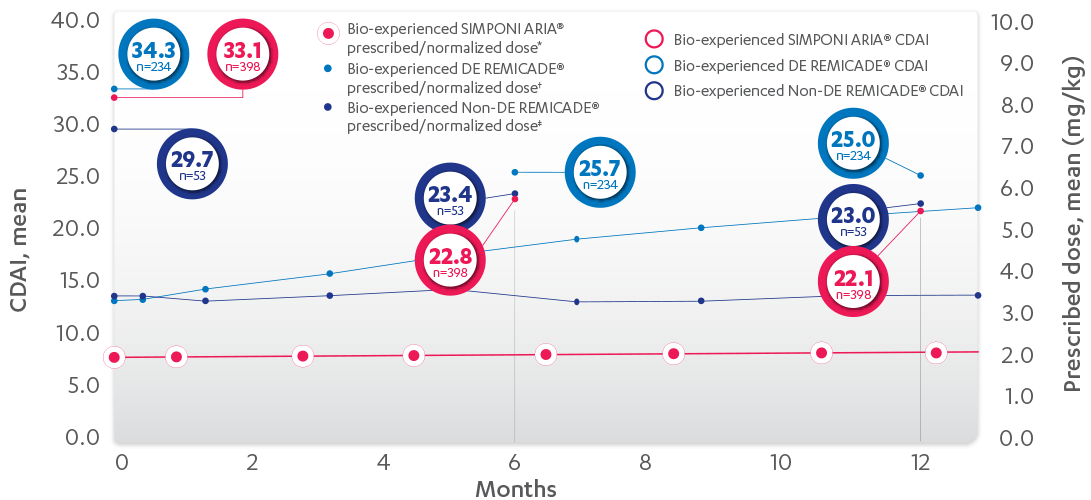
Patients Per Infusion
Results are not intended for direct comparison with Phase 3 clinical trials because the real-world studies were observational trials. There have been no head-to-head randomized clinical trials comparing the safety and efficacy of SIMPONI ARIA® vs REMICADE®.
SIMPONI ARIA® IPTW imputed mean change† from baseline at 6 months=–10.3 and 12 months=–11.0. DE= at least 1 post-baseline normalized dose > baseline dose. REMICADE® DE IPTW imputed mean change† from baseline at 6 months=–8.6 and 12 months=–9.3. REMICADE® non-DE imputed mean change† from baseline at 6 months=–6.3 and 12 months=–6.7.
*LOCF with treatment failure rules.
†Means with weight on IPTW propensity score.
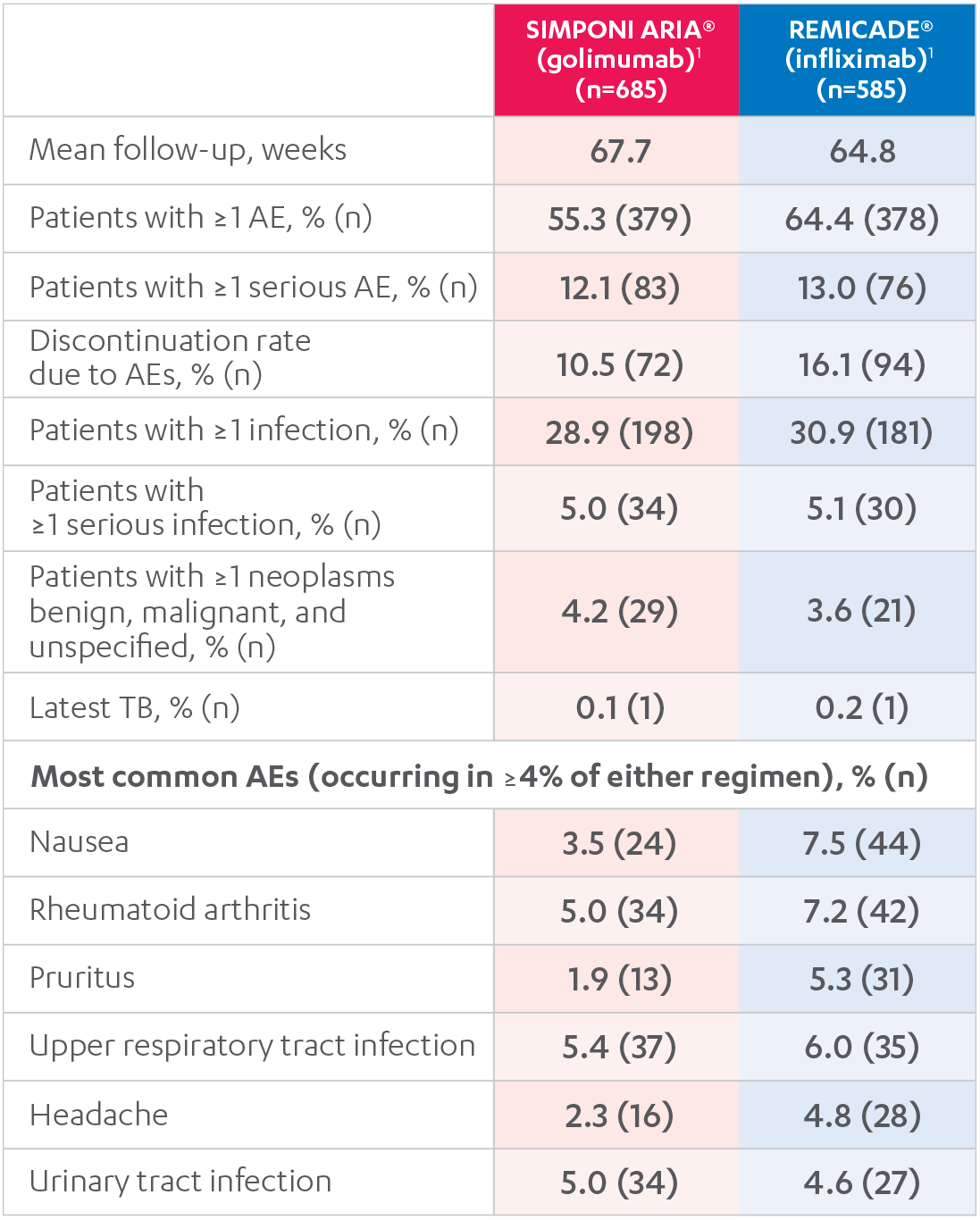
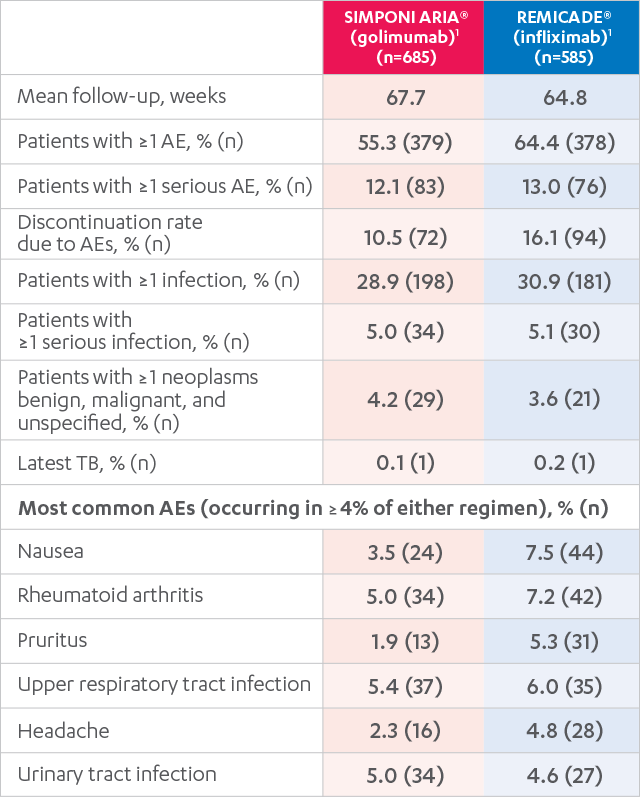
ADVERSE EVENTS, 52 WEEKS1
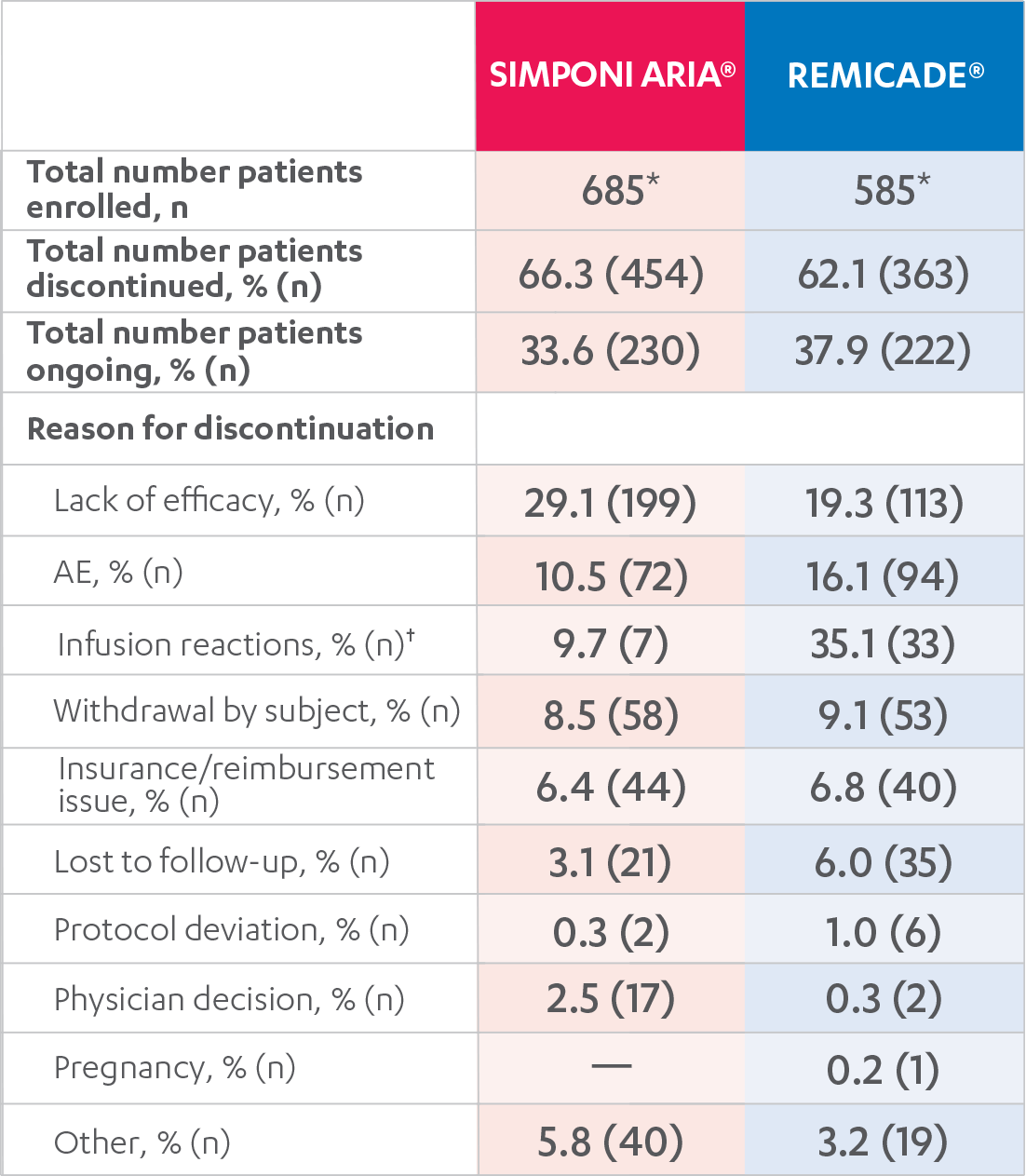
DISPOSITIONS AND DISCONTINUATIONS, 52 WEEKS
FULL ANALYSIS SET1,2
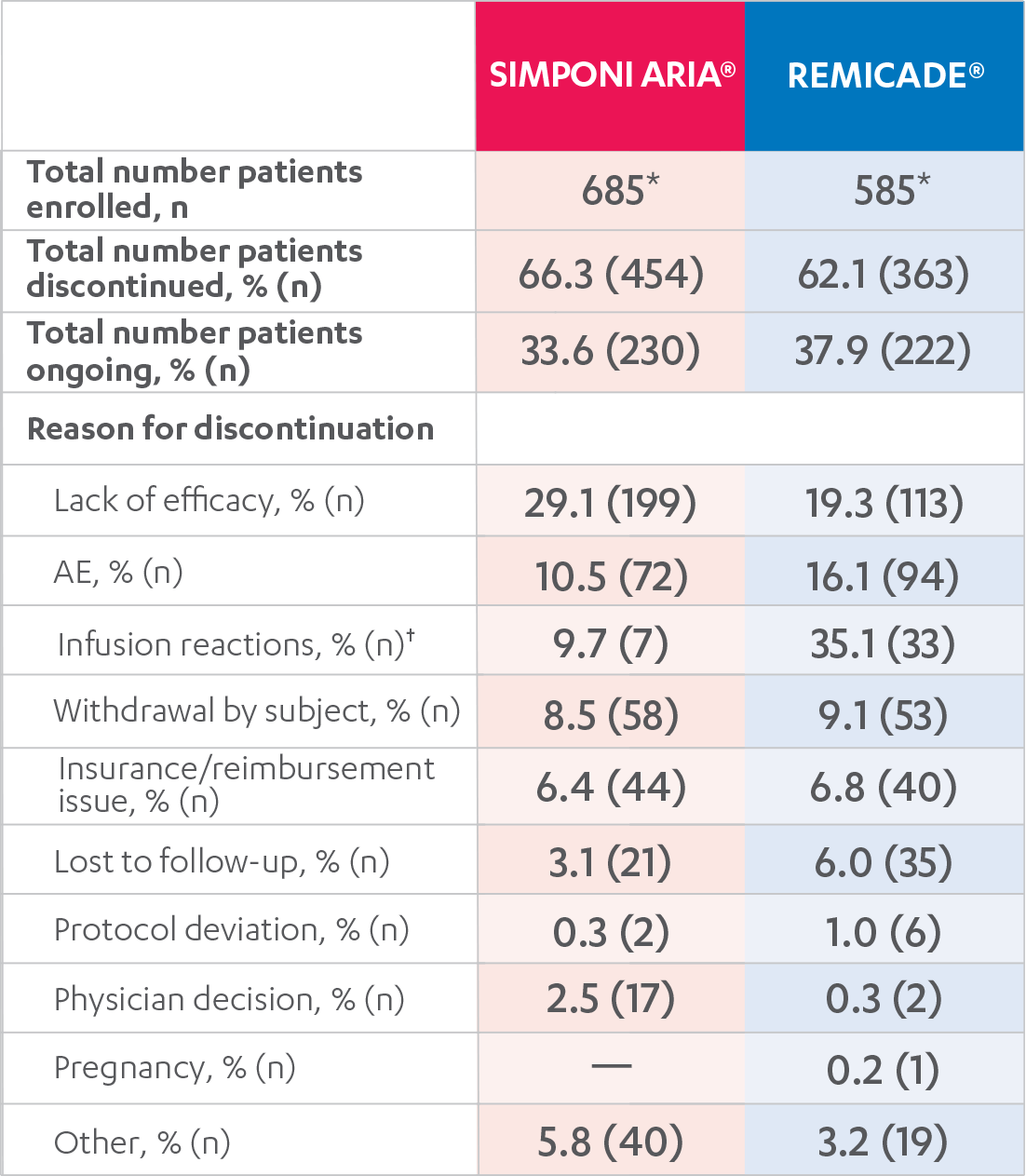
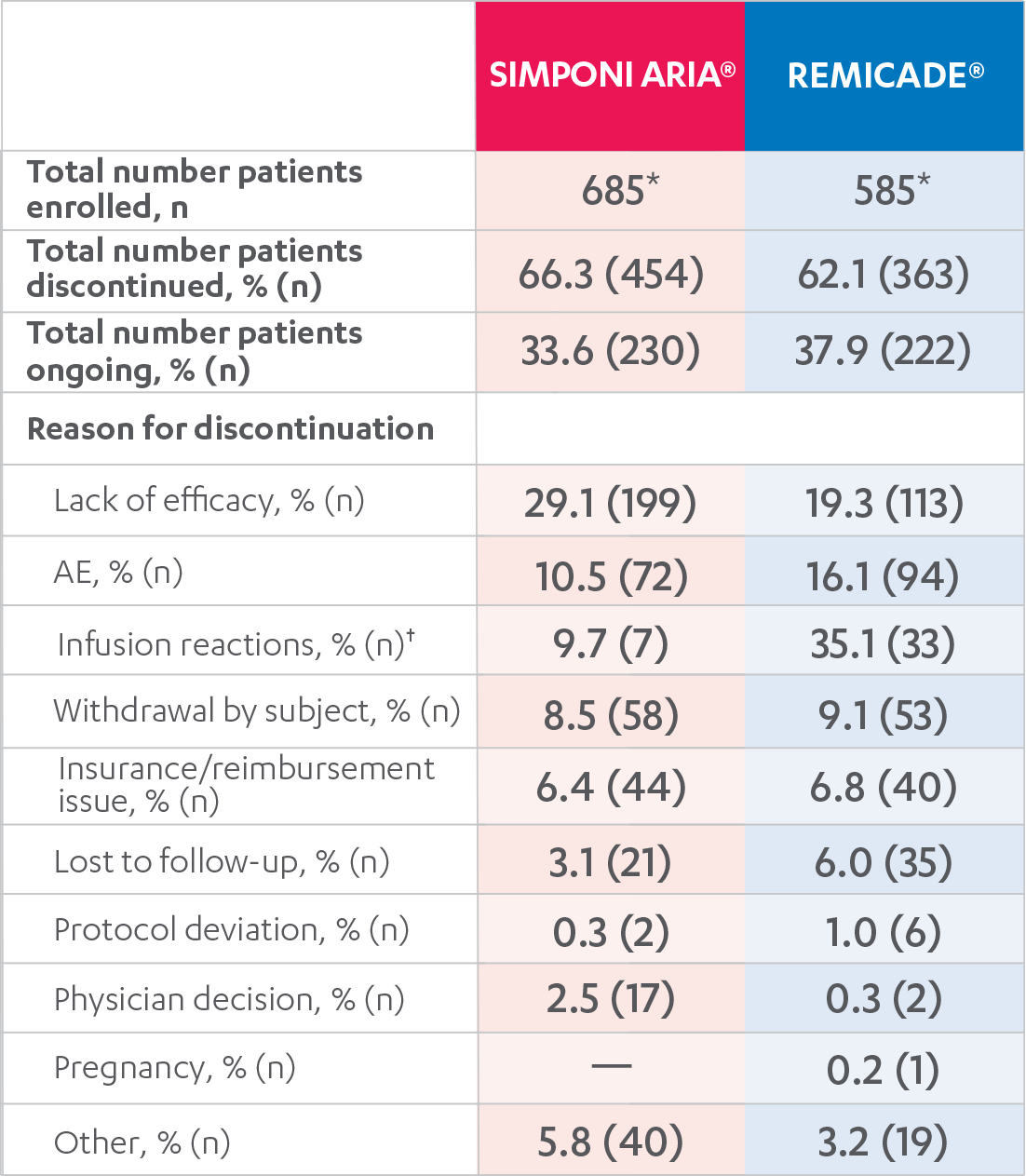
*One SIMPONI ARIA® patient completed the planned 3 years of treatment (as of February 1, 2019).
†Expressed as a percent of AEs.
Results are not intended for direct comparison with Phase 3 clinical trials because the real-world studies were observational trials. There have been no head-to-head randomized clinical trials comparing the safety and efficacy of SIMPONI ARIA® vs REMICADE®.
IMPORTANT SAFETY INFORMATION
SERIOUS INFECTIONS
Patients treated with SIMPONI ARIA® (golimumab) are at increased risk for developing serious infections that may lead to hospitalization or death. Most patients who developed these infections were taking concomitant immunosuppressants such as methotrexate or corticosteroids. Discontinue SIMPONI ARIA® if a patient develops a serious infection.
Reported infections with TNF blockers, of which SIMPONI ARIA® is a member, include:
- Active tuberculosis (TB), including reactivation of latent TB. Patients frequently presented with disseminated or extrapulmonary disease. Test patients for latent TB before SIMPONI ARIA® use and during therapy. Initiate treatment for latent infection prior to SIMPONI ARIA® use.
- Invasive fungal infections, including histoplasmosis, coccidioidomycosis, candidiasis, aspergillosis, blastomycosis, and pneumocystosis. Patients with histoplasmosis or other invasive fungal infections may present with disseminated, rather than localized, disease. Consider empiric anti-fungal therapy in patients at risk for invasive fungal infections who develop severe systemic illness.
- Bacterial, viral, and other infections due to opportunistic pathogens, including Legionella and Listeria.
Consider the risks and benefits of treatment with SIMPONI ARIA® prior to initiating therapy in patients with chronic or recurrent infection. Do not start SIMPONI ARIA® in patients with clinically important active infections, including localized infections. Closely monitor patients for the development of signs and symptoms of infection during and after treatment with SIMPONI ARIA®, including the possible development of TB in patients who tested negative for latent TB infection prior to initiating therapy, who are on treatment for latent TB, or who were previously treated for TB infection.
Risk of infection may be higher in patients greater than 65 years of age, patients with co-morbid conditions and/or patients taking concomitant immunosuppressant therapy. Other serious infections observed in patients treated with SIMPONI ARIA® included sepsis, pneumonia, cellulitis, and abscess.
MALIGNANCIES
cp-51207v3Malignancies, some fatal, have been reported in children, adolescents, and young adult patients treated with golimumab. Approximately half the cases were lymphomas, including Hodgkin’s and non-Hodgkin’s lymphoma. The other cases represented a variety of malignancies, including rare malignancies usually associated with immunosuppression and malignancies not usually observed in children or adolescents. Malignancies occurred after a median of 30 months after the first dose of therapy. Most of the patients were receiving concomitant immunosuppressants.
In the controlled portions of clinical trials of TNF blockers including the subcutaneous formulation of golimumab, more cases of lymphoma have been observed among patients receiving anti-TNF treatment compared with patients in the control groups. In clinical trials, the incidence of malignancies other than lymphoma and non-melanoma skin cancer per 100 patient-years of follow-up was 0.56 (95% CI: 0.01, 3.11) in the SIMPONI ARIA® group compared with an incidence of 0 (95% CI: 0.00, 3.79) in the placebo group. Cases of acute and chronic leukemia have been reported with TNF-blocker use, including SIMPONI ARIA®. The risks and benefits of TNF-blocker therapy should be considered prior to initiating therapy in patients with a known malignancy or who develop a malignancy.
Postmarketing cases of hepatosplenic T-cell lymphoma (HSTCL), a rare type of T-cell lymphoma, have been reported in patients treated with TNF blockers. These cases have had a very aggressive disease course and have been fatal. Nearly all reported cases have occurred in patients with Crohn’s disease or ulcerative colitis, and the majority were in adolescent and young adult males. Almost all of these patients had received treatment with azathioprine or 6-mercaptopurine concomitantly with a TNF blocker at or prior to diagnosis. A risk for the development for HSTCL in patients treated with TNF blockers cannot be excluded.
Melanoma and Merkel cell carcinoma have been reported in patients treated with TNF-blocking agents, including SIMPONI ARIA®. Periodic skin examination is recommended for all patients, particularly those with risk factors for skin cancer.
HEPATITIS B REACTIVATION
The use of TNF blockers, of which SIMPONI ARIA® is a member, has been associated with reactivation of hepatitis B virus (HBV) in patients who are chronic hepatitis B carriers. In some instances, HBV reactivation occurring in conjunction with TNF-blocker therapy has been fatal. The majority of these reports have occurred in patients who received concomitant immunosuppressants.
All patients should be tested for HBV infection before initiating TNF-blocker therapy. For patients who test positive for hepatitis B surface antigen, consult a physician with expertise in the treatment of hepatitis B before initiating TNF-blocker therapy. Exercise caution when prescribing SIMPONI ARIA® for patients identified as carriers of HBV and closely monitor for active HBV infection during and following termination of therapy with SIMPONI ARIA®. Discontinue SIMPONI ARIA® in patients who develop HBV reactivation, and initiate antiviral therapy with appropriate supportive treatment. Exercise caution when considering resumption of SIMPONI ARIA®, and monitor patients closely.
CONGESTIVE HEART FAILURE
Cases of worsening congestive heart failure (CHF) and new-onset CHF have been reported with TNF blockers, including SIMPONI ARIA®. Some cases had a fatal outcome. Exercise caution in CHF patients receiving SIMPONI ARIA® and monitor them closely during therapy. Discontinue SIMPONI ARIA® if new or worsening symptoms of heart failure appear.
DEMYELINATING DISORDERS
Use of TNF blockers, including SIMPONI ARIA®, has been associated with rare cases of new-onset or exacerbation of demyelinating disorders, including multiple sclerosis (MS) and Guillain-Barré syndrome. Cases of central demyelination, MS, optic neuritis, and peripheral demyelinating polyneuropathy have rarely been reported in patients treated with golimumab. Exercise caution in considering the use of SIMPONI ARIA® in patients with these disorders. Consider discontinuation if these disorders develop.
AUTOIMMUNITY
Treatment with TNF blockers, including SIMPONI ARIA®, may result in the formation of antinuclear antibodies. Rarely, treatment with TNF blockers may result in a lupus-like syndrome. Discontinue treatment if symptoms of a lupus-like syndrome develop.
USE WITH OTHER DRUGS
The concomitant use of a TNF blocker and abatacept or anakinra was associated with a higher risk of serious infections, therefore the use of SIMPONI ARIA® in combination with these products is not recommended. Care should be taken when switching from one biologic to another since overlapping biological activity may further increase the risk of infection. A higher rate of serious infections has also been observed in RA patients treated with rituximab who received subsequent treatment with a TNF blocker. The concomitant use of SIMPONI ARIA® with biologics approved to treat RA is not recommended because of the possibility of an increased risk of infection.
HEMATOLOGIC CYTOPENIAS
There have been reports of pancytopenia, leukopenia, neutropenia, agranulocytosis, aplastic anemia, and thrombocytopenia in patients receiving SIMPONI ARIA®. Exercise caution when using SIMPONI ARIA® in patients who have or had significant cytopenias.
VACCINATIONS/THERAPEUTIC INFECTIOUS AGENTS
Live vaccines or therapeutic infectious agents should not be given with SIMPONI ARIA® due to the possibility of clinical infections, including disseminated infections.
Update vaccinations prior to initiation of treatment in accordance with current vaccination guidelines. Advise patients to discuss with the physician before seeking any immunizations. At least a 6-month waiting period following birth is recommended before the administration of any live vaccine to infants exposed in utero to SIMPONI ARIA®.
HYPERSENSITIVITY REACTIONS
Serious systemic hypersensitivity reactions (including anaphylaxis) have been reported following administration of the subcutaneous formulation of golimumab and SIMPONI ARIA®, some occurring after the first dose. Hypersensitivity reactions including hives, pruritus, dyspnea, and nausea, were reported in association with infusions of SIMPONI ARIA®. If an anaphylactic or other serious allergic reaction occurs, discontinue SIMPONI ARIA® immediately and institute appropriate therapy.
ADVERSE REACTIONS
The most serious adverse reactions were serious infections and malignancies.
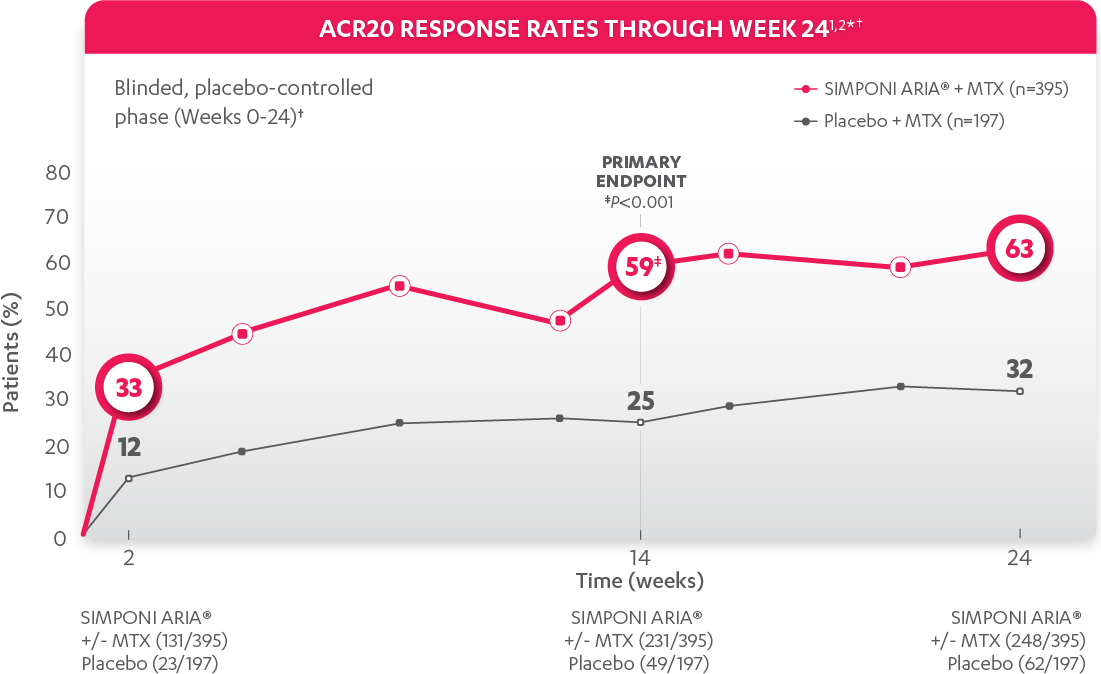
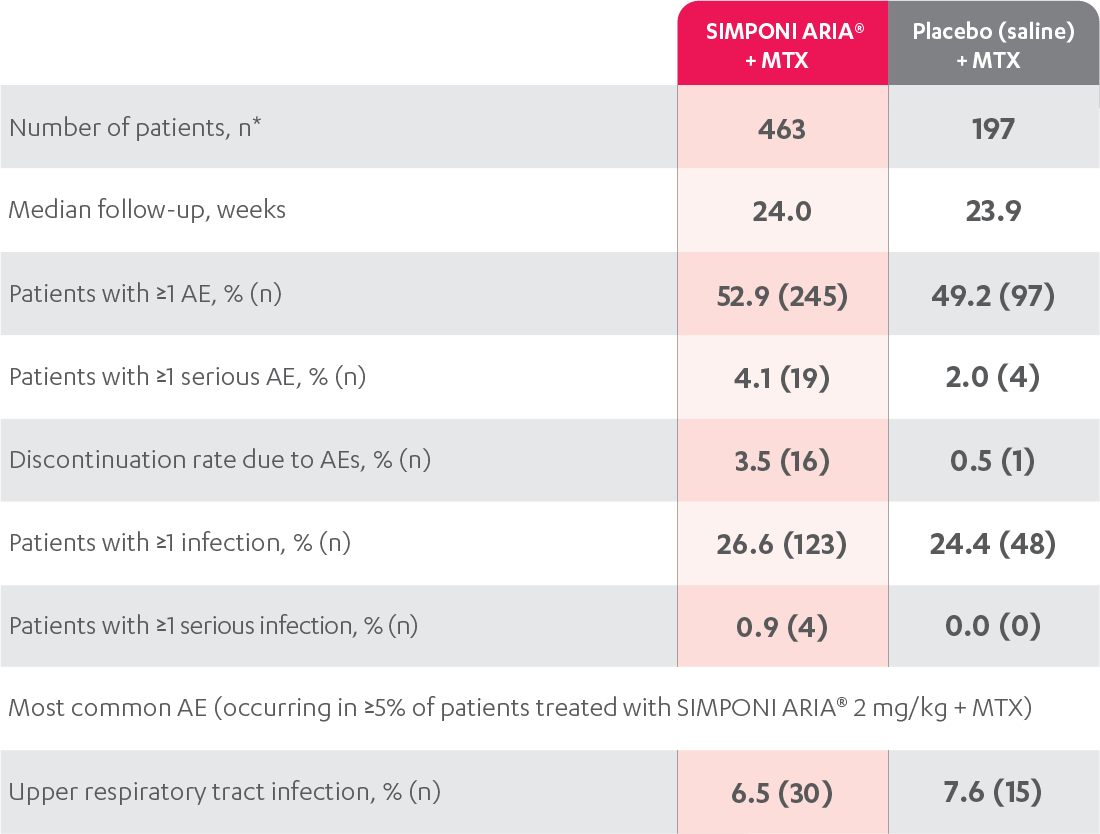
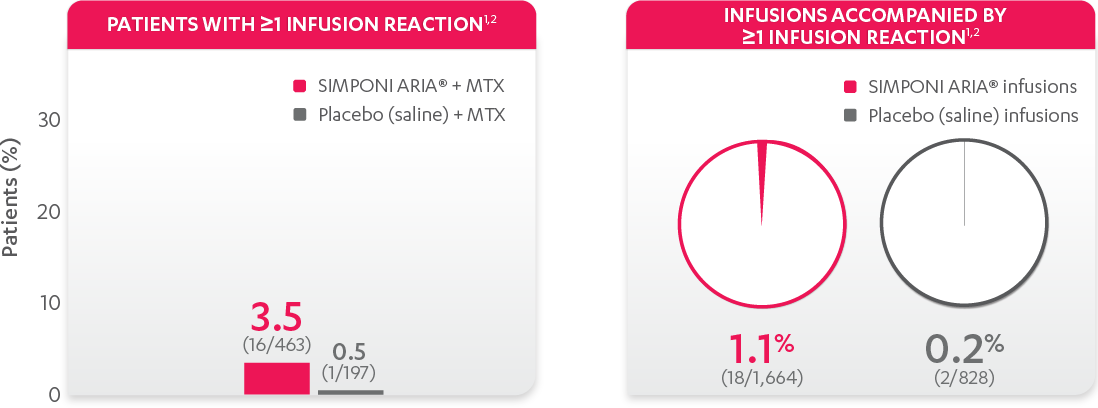
The most common adverse reactions (incidence ≥3%) reported in clinical trials were: upper respiratory tract infection, alanine aminotransferase increase, viral infection, aspartate aminotransferase increase, neutrophil count decrease, bronchitis, hypertension, and rash. In the controlled phase of Trial RA, the rate of infusions associated with an infusion reaction was reported in 1.1% of SIMPONI ARIA® infusions compared with 0.2% of infusions in the control group.
The adverse reactions observed in pediatric patients with polyarticular Juvenile Idiopathic Arthritis (pJIA) were consistent with the established safety profile of SIMPONI ARIA® in adult patients with RA and PsA.
Please see the full Prescribing Information and Medication Guide for SIMPONI ARIA®. Provide the Medication Guide to your patients and encourage discussion.
Indication for
SIMPONI ARIA® (golimumab)
INDICATION
SIMPONI ARIA® is indicated for the treatment of adults with moderately to severely active rheumatoid arthritis (RA) in combination with methotrexate (MTX).
IMPORTANT SAFETY INFORMATION
SERIOUS INFECTIONS
Patients treated with REMICADE® (infliximab) are at increased risk for developing serious infections that may lead to hospitalization or death. Most patients who developed these infections were taking concomitant immunosuppressants such as methotrexate or corticosteroids. Discontinue REMICADE® if a patient develops a serious infection or sepsis.
Reported infections include:
- Active tuberculosis (TB), including reactivation of latent TB. Patients frequently presented with disseminated or extrapulmonary disease. Patients should be tested for latent TB before and during treatment with REMICADE®.1,2 Treatment for latent infection should be initiated prior to treatment with REMICADE®.
- Invasive fungal infections, including histoplasmosis, coccidioidomycosis, candidiasis, aspergillosis, blastomycosis, pneumocystosis, and cryptococcosis. Patients may present with disseminated, rather than localized, disease. Empiric anti-fungal therapy should be considered in patients at risk for invasive fungal infections who develop severe systemic illness.
- Bacterial, viral, and other infections due to opportunistic pathogens, including Legionella, Listeria, and Salmonella.
The risks and benefits of treatment with REMICADE® should be carefully considered prior to initiating therapy in patients with chronic or recurrent infection. Closely monitor patients for the development of signs and symptoms of infection during and after treatment with REMICADE®, including the possible development of TB in patients who tested negative for latent TB infection prior to initiating therapy, who are on treatment for latent TB, or who were previously treated for TB infection.
Risk of infection may be higher in patients greater than 65 years of age, pediatric patients, patients with co-morbid conditions and/or patients taking concomitant immunosuppressant therapy. In clinical trials, other serious infections observed in patients treated with REMICADE® included pneumonia, cellulitis, abscess, and skin ulceration.
MALIGNANCIES
cp-62063v2Lymphoma and other malignancies, some fatal, have been reported in children and adolescent patients treated with TNF blockers, including REMICADE®. Approximately half of these cases were lymphomas, including Hodgkin’s and non-Hodgkin’s lymphoma. The other cases represented a variety of malignancies, including rare malignancies that are usually associated with immunosuppression and malignancies that are not usually observed in children and adolescents. The malignancies occurred after a median of 30 months after the first dose of therapy. Most of the patients were receiving concomitant immunosuppressants.
Postmarketing cases of hepatosplenic T-cell lymphoma, a rare type of T-cell lymphoma, have been reported in patients treated with TNF blockers, including REMICADE®. These cases have had a very aggressive disease course and have been fatal. The majority of reported REMICADE® cases have occurred in patients with Crohn’s disease or ulcerative colitis and most were in adolescent and young adult males. Almost all of these patients had received treatment with azathioprine or 6-mercaptopurine concomitantly with REMICADE® at or prior to diagnosis. Carefully assess the risks and benefits of treatment with REMICADE®, especially in these patient types.
In clinical trials of all TNF blockers, more cases of lymphoma were observed compared with controls and the expected rate in the general population. However, patients with Crohn’s disease, rheumatoid arthritis, or plaque psoriasis may be at higher risk for developing lymphoma. In clinical trials of some TNF blockers, including REMICADE®, more cases of other malignancies were observed compared with controls. The rate of these malignancies among patients treated with REMICADE® was similar to that expected in the general population whereas the rate in control patients was lower than expected. Cases of acute and chronic leukemia have been reported with postmarketing TNF-blocker use. As the potential role of TNF blockers in the development of malignancies is not known, caution should be exercised when considering treatment of patients with a current or a past history of malignancy or other risk factors such as chronic obstructive pulmonary disease (COPD).
Melanoma and Merkel cell carcinoma have been reported in patients treated with TNF‑blocker therapy, including REMICADE®. Periodic skin examination is recommended for all patients, particularly those with risk factors for skin cancer.
A population-based retrospective cohort study found a 2- to 3-fold increase in the incidence of invasive cervical cancer in women with rheumatoid arthritis treated with REMICADE® compared to biologics-naïve patients or the general population, particularly those over 60 years of age. A causal relationship between REMICADE® and cervical cancer cannot be excluded. Periodic screening should continue in women treated with REMICADE®.
CONTRAINDICATIONS
The use of REMICADE® at doses >5 mg/kg is contraindicated in patients with moderate or severe heart failure. REMICADE® is contraindicated in patients with a previous severe hypersensitivity reaction to infliximab or any of the inactive ingredients of REMICADE® or any murine proteins (severe hypersensitivity reactions have included anaphylaxis, hypotension, and serum sickness).
HEPATITIS B REACTIVATION
TNF blockers, including REMICADE®, have been associated with reactivation of hepatitis B virus (HBV) in patients who are chronic carriers. Some cases were fatal. Patients should be tested for HBV infection before initiating REMICADE®. For patients who test positive, consult a physician with expertise in the treatment of hepatitis B. Exercise caution when prescribing REMICADE® for patients identified as carriers of HBV and monitor closely for active HBV infection during and following termination of therapy with REMICADE®. Discontinue REMICADE® in patients who develop HBV reactivation and initiate antiviral therapy with appropriate supportive treatment. Exercise caution when considering resumption of REMICADE® and monitor patients closely.
HEPATOTOXICITY
Severe hepatic reactions, including acute liver failure, jaundice, hepatitis, and cholestasis have been reported in patients receiving REMICADE® postmarketing. Some cases were fatal or required liver transplant. Aminotransferase elevations were not noted prior to discovery of liver injury in many cases. Patients with symptoms or signs of liver dysfunction should be evaluated for evidence of liver injury. If jaundice and/or marked liver enzyme elevations (eg, ≥5 times the upper limit of normal) develop, REMICADE® should be discontinued, and a thorough investigation of the abnormality should be undertaken.
HEART FAILURE
In a randomized, placebo-controlled study in patients with moderate or severe heart failure (NYHA Functional Class III/IV), higher mortality rates and a higher risk of hospitalization were observed at Week 28 at a dose of 10 mg/kg and higher rates of cardiovascular events were observed at both 5 mg/kg and 10 mg/kg. There have been postmarketing reports of new onset and worsening heart failure, with and without identifiable precipitating factors. Patients with moderate or severe heart failure taking REMICADE® (≤5 mg/kg) or patients with mild heart failure should be closely monitored and treatment should be discontinued if new or worsening symptoms appear.
HEMATOLOGIC EVENTS
Cases of leukopenia, neutropenia, thrombocytopenia, and pancytopenia (some fatal) have been reported. The causal relationship to REMICADE® therapy remains unclear. Exercise caution in patients who have ongoing or a history of significant hematologic abnormalities. Advise patients to seek immediate medical attention if they develop signs and symptoms of blood dyscrasias or infection. Consider discontinuation of REMICADE® in patients who develop significant hematologic abnormalities.
HYPERSENSITIVITY
REMICADE® has been associated with hypersensitivity reactions that differ in their time of onset. Anaphylaxis, acute urticaria, dyspnea, and hypotension have occurred in association with infusions of REMICADE®. Medications for the treatment of hypersensitivity reactions should be available.
CARDIOVASCULAR AND CEREBROVASCULAR REACTIONS DURING AND AFTER INFUSION
Serious cerebrovascular accidents, myocardial ischemia/infarction (some fatal), hypotension, hypertension, and arrhythmias have been reported during and within 24 hours of initiation of REMICADE® infusion. Cases of transient visual loss have been reported during or within 2 hours of REMICADE® infusion. Monitor patients during infusion and if a serious reaction occurs, discontinue infusion. Manage reactions according to signs and symptoms.
NEUROLOGIC EVENTS
TNF blockers, including REMICADE®, have been associated with CNS manifestation of systemic vasculitis, seizure, and new onset or exacerbation of CNS demyelinating disorders, including multiple sclerosis and optic neuritis, and peripheral demyelinating disorders, including Guillain- Barré syndrome. Exercise caution when considering REMICADE® in patients with these disorders and consider discontinuation if these disorders develop.
CONCURRENT ADMINISTRATION WITH OTHER BIOLOGICS
Concurrent use of REMICADE® with anakinra, abatacept, tocilizumab, or other biologics used to treat the same conditions as REMICADE® is not recommended because of the possibility of an increased risk of infection. Care should be taken when switching from one biologic to another, since overlapping biological activity may further increase the risk of infection.
AUTOIMMUNITY
Treatment with REMICADE® may result in the formation of autoantibodies and in the development of a lupus-like syndrome. Discontinue treatment if symptoms of a lupus-like syndrome develop.
VACCINATIONS AND USE OF LIVE VACCINES/THERAPEUTIC INFECTIOUS AGENTS
Prior to initiating REMICADE®, update vaccinations in accordance with current vaccination guidelines. Live vaccines or therapeutic infectious agents should not be given with REMICADE® due to the possibility of clinical infections, including disseminated infections.
At least a 6-month waiting period following birth is recommended before the administration of any live vaccine to infants exposed in utero to REMICADE®.
ADVERSE REACTIONS
In clinical trials, the most common adverse reactions occurring in >10% of REMICADE®-treated patients included infections (eg, upper respiratory, sinusitis, and pharyngitis), infusion-related reactions, headache, and abdominal pain.
For more information, please see the full Prescribing Information and Medication Guide for REMICADE®. Provide the Medication Guide to your patients and encourage discussion.
Indication for
REMICADE® (infliximab)
INDICATION
REMICADE®, in combination with methotrexate, is indicated for reducing signs and symptoms, inhibiting the progression of structural damage, and improving physical function in adult patients with moderately to severely active rheumatoid arthritis (RA).
©Janssen Biotech, Inc. 2011-2020. All rights reserved. cp-71889v5 Last Updated: October 2020
This site is published by Janssen Biotech, Inc., which is solely responsible for its contents. This site is intended for use by healthcare professionals of the United States and Puerto Rico. Janssen Biotech, Inc., recognizes that the Internet is a global communications medium; however, laws, regulatory requirements, and medical practices for pharmaceutical products vary from country to country. The Prescribing Information included here may not be appropriate for use outside the United States and Puerto Rico. Third party trademarks used herein are trademarks of their respective owners.
SIMPONIARIAHCP.com|REMICADEHCP.com
SIMPONI ARIA® Prescribing Information|REMICADE® Prescribing Information
Medical Information Center
Indication for
SIMPONI ARIA® (golimumab)
INDICATION
SIMPONI ARIA® is indicated for the treatment of adults with moderately to severely active rheumatoid arthritis (RA) in combination with methotrexate (MTX).
IMPORTANT SAFETY INFORMATION
SERIOUS INFECTIONS
Patients treated with SIMPONI ARIA® (golimumab) are at increased risk for developing serious infections that may lead to hospitalization or death. Most patients who developed these infections were taking concomitant immunosuppressants such as methotrexate or corticosteroids. Discontinue SIMPONI ARIA® if a patient develops a serious infection.
Reported infections with TNF blockers, of which SIMPONI ARIA® is a member, include:
- Active tuberculosis (TB), including reactivation of latent TB. Patients frequently presented with disseminated or extrapulmonary disease. Test patients for latent TB before SIMPONI ARIA® use and during therapy. Initiate treatment for latent infection prior to SIMPONI ARIA® use.
- Invasive fungal infections, including histoplasmosis, coccidioidomycosis, candidiasis, aspergillosis, blastomycosis, and pneumocystosis. Patients with histoplasmosis or other invasive fungal infections may present with disseminated, rather than localized, disease. Consider empiric anti-fungal therapy in patients at risk for invasive fungal infections who develop severe systemic illness.
- Bacterial, viral, and other infections due to opportunistic pathogens, including Legionella and Listeria.
Consider the risks and benefits of treatment with SIMPONI ARIA® prior to initiating therapy in patients with chronic or recurrent infection. Do not start SIMPONI ARIA® in patients with clinically important active infections, including localized infections. Closely monitor patients for the development of signs and symptoms of infection during and after treatment with SIMPONI ARIA®, including the possible development of TB in patients who tested negative for latent TB infection prior to initiating therapy, who are on treatment for latent TB, or who were previously treated for TB infection.
Risk of infection may be higher in patients greater than 65 years of age, patients with co-morbid conditions and/or patients taking concomitant immunosuppressant therapy. Other serious infections observed in patients treated with SIMPONI ARIA® included sepsis, pneumonia, cellulitis, and abscess.
MALIGNANCIES
cp-51207v3Malignancies, some fatal, have been reported in children, adolescents, and young adult patients treated with golimumab. Approximately half the cases were lymphomas, including Hodgkin’s and non-Hodgkin’s lymphoma. The other cases represented a variety of malignancies, including rare malignancies usually associated with immunosuppression and malignancies not usually observed in children or adolescents. Malignancies occurred after a median of 30 months after the first dose of therapy. Most of the patients were receiving concomitant immunosuppressants.
In the controlled portions of clinical trials of TNF blockers including the subcutaneous formulation of golimumab, more cases of lymphoma have been observed among patients receiving anti-TNF treatment compared with patients in the control groups. In clinical trials, the incidence of malignancies other than lymphoma and non-melanoma skin cancer per 100 patient-years of follow-up was 0.56 (95% CI: 0.01, 3.11) in the SIMPONI ARIA® group compared with an incidence of 0 (95% CI: 0.00, 3.79) in the placebo group. Cases of acute and chronic leukemia have been reported with TNF-blocker use, including SIMPONI ARIA®. The risks and benefits of TNF-blocker therapy should be considered prior to initiating therapy in patients with a known malignancy or who develop a malignancy.
Postmarketing cases of hepatosplenic T-cell lymphoma (HSTCL), a rare type of T-cell lymphoma, have been reported in patients treated with TNF blockers. These cases have had a very aggressive disease course and have been fatal. Nearly all reported cases have occurred in patients with Crohn’s disease or ulcerative colitis, and the majority were in adolescent and young adult males. Almost all of these patients had received treatment with azathioprine or 6-mercaptopurine concomitantly with a TNF blocker at or prior to diagnosis. A risk for the development for HSTCL in patients treated with TNF blockers cannot be excluded.
Melanoma and Merkel cell carcinoma have been reported in patients treated with TNF-blocking agents, including SIMPONI ARIA®. Periodic skin examination is recommended for all patients, particularly those with risk factors for skin cancer.
HEPATITIS B REACTIVATION
The use of TNF blockers, of which SIMPONI ARIA® is a member, has been associated with reactivation of hepatitis B virus (HBV) in patients who are chronic hepatitis B carriers. In some instances, HBV reactivation occurring in conjunction with TNF-blocker therapy has been fatal. The majority of these reports have occurred in patients who received concomitant immunosuppressants.
All patients should be tested for HBV infection before initiating TNF-blocker therapy. For patients who test positive for hepatitis B surface antigen, consult a physician with expertise in the treatment of hepatitis B before initiating TNF-blocker therapy. Exercise caution when prescribing SIMPONI ARIA® for patients identified as carriers of HBV and closely monitor for active HBV infection during and following termination of therapy with SIMPONI ARIA®. Discontinue SIMPONI ARIA® in patients who develop HBV reactivation, and initiate antiviral therapy with appropriate supportive treatment. Exercise caution when considering resumption of SIMPONI ARIA®, and monitor patients closely.
CONGESTIVE HEART FAILURE
Cases of worsening congestive heart failure (CHF) and new-onset CHF have been reported with TNF blockers, including SIMPONI ARIA®. Some cases had a fatal outcome. Exercise caution in CHF patients receiving SIMPONI ARIA® and monitor them closely during therapy. Discontinue SIMPONI ARIA® if new or worsening symptoms of heart failure appear.
DEMYELINATING DISORDERS
Use of TNF blockers, including SIMPONI ARIA®, has been associated with rare cases of new-onset or exacerbation of demyelinating disorders, including multiple sclerosis (MS) and Guillain-Barré syndrome. Cases of central demyelination, MS, optic neuritis, and peripheral demyelinating polyneuropathy have rarely been reported in patients treated with golimumab. Exercise caution in considering the use of SIMPONI ARIA® in patients with these disorders. Consider discontinuation if these disorders develop.
AUTOIMMUNITY
Treatment with TNF blockers, including SIMPONI ARIA®, may result in the formation of antinuclear antibodies. Rarely, treatment with TNF blockers may result in a lupus-like syndrome. Discontinue treatment if symptoms of a lupus-like syndrome develop.
USE WITH OTHER DRUGS
The concomitant use of a TNF blocker and abatacept or anakinra was associated with a higher risk of serious infections, therefore the use of SIMPONI ARIA® in combination with these products is not recommended. Care should be taken when switching from one biologic to another since overlapping biological activity may further increase the risk of infection. A higher rate of serious infections has also been observed in RA patients treated with rituximab who received subsequent treatment with a TNF blocker. The concomitant use of SIMPONI ARIA® with biologics approved to treat RA is not recommended because of the possibility of an increased risk of infection.
HEMATOLOGIC CYTOPENIAS
There have been reports of pancytopenia, leukopenia, neutropenia, agranulocytosis, aplastic anemia, and thrombocytopenia in patients receiving SIMPONI ARIA®. Exercise caution when using SIMPONI ARIA® in patients who have or had significant cytopenias.
VACCINATIONS/THERAPEUTIC INFECTIOUS AGENTS
Live vaccines or therapeutic infectious agents should not be given with SIMPONI ARIA® due to the possibility of clinical infections, including disseminated infections.
Update vaccinations prior to initiation of treatment in accordance with current vaccination guidelines. Advise patients to discuss with the physician before seeking any immunizations. At least a 6-month waiting period following birth is recommended before the administration of any live vaccine to infants exposed in utero to SIMPONI ARIA®.
HYPERSENSITIVITY REACTIONS
Serious systemic hypersensitivity reactions (including anaphylaxis) have been reported following administration of the subcutaneous formulation of golimumab and SIMPONI ARIA®, some occurring after the first dose. Hypersensitivity reactions including hives, pruritus, dyspnea, and nausea, were reported in association with infusions of SIMPONI ARIA®. If an anaphylactic or other serious allergic reaction occurs, discontinue SIMPONI ARIA® immediately and institute appropriate therapy.
ADVERSE REACTIONS
The most serious adverse reactions were serious infections and malignancies.
The most common adverse reactions (incidence ≥3%) reported in clinical trials were: upper respiratory tract infection, alanine aminotransferase increase, viral infection, aspartate aminotransferase increase, neutrophil count decrease, bronchitis, hypertension, and rash. In the controlled phase of Trial RA, the rate of infusions associated with an infusion reaction was reported in 1.1% of SIMPONI ARIA® infusions compared with 0.2% of infusions in the control group.
The adverse reactions observed in pediatric patients with polyarticular Juvenile Idiopathic Arthritis (pJIA) were consistent with the established safety profile of SIMPONI ARIA® in adult patients with RA and PsA.
Please see the full Prescribing Information and Medication Guide for SIMPONI ARIA®. Provide the Medication Guide to your patients and encourage discussion.

Indication for
REMICADE® (infliximab)
INDICATION
REMICADE®, in combination with methotrexate, is indicated for reducing signs and symptoms, inhibiting the progression of structural damage, and improving physical function in adult patients with moderately to severely active rheumatoid arthritis (RA).
IMPORTANT SAFETY INFORMATION
SERIOUS INFECTIONS
Patients treated with REMICADE® (infliximab) are at increased risk for developing serious infections that may lead to hospitalization or death. Most patients who developed these infections were taking concomitant immunosuppressants such as methotrexate or corticosteroids. Discontinue REMICADE® if a patient develops a serious infection or sepsis.
Reported infections include:
- Active tuberculosis (TB), including reactivation of latent TB. Patients frequently presented with disseminated or extrapulmonary disease. Patients should be tested for latent TB before and during treatment with REMICADE®.1,2 Treatment for latent infection should be initiated prior to treatment with REMICADE®.
- Invasive fungal infections, including histoplasmosis, coccidioidomycosis, candidiasis, aspergillosis, blastomycosis, pneumocystosis, and cryptococcosis. Patients may present with disseminated, rather than localized, disease. Empiric anti-fungal therapy should be considered in patients at risk for invasive fungal infections who develop severe systemic illness.
- Bacterial, viral, and other infections due to opportunistic pathogens, including Legionella, Listeria, and Salmonella.
The risks and benefits of treatment with REMICADE® should be carefully considered prior to initiating therapy in patients with chronic or recurrent infection. Closely monitor patients for the development of signs and symptoms of infection during and after treatment with REMICADE®, including the possible development of TB in patients who tested negative for latent TB infection prior to initiating therapy, who are on treatment for latent TB, or who were previously treated for TB infection.
Risk of infection may be higher in patients greater than 65 years of age, pediatric patients, patients with co-morbid conditions and/or patients taking concomitant immunosuppressant therapy. In clinical trials, other serious infections observed in patients treated with REMICADE® included pneumonia, cellulitis, abscess, and skin ulceration.
MALIGNANCIES
cp-62063v2Lymphoma and other malignancies, some fatal, have been reported in children and adolescent patients treated with TNF blockers, including REMICADE®. Approximately half of these cases were lymphomas, including Hodgkin’s and non-Hodgkin’s lymphoma. The other cases represented a variety of malignancies, including rare malignancies that are usually associated with immunosuppression and malignancies that are not usually observed in children and adolescents. The malignancies occurred after a median of 30 months after the first dose of therapy. Most of the patients were receiving concomitant immunosuppressants.
Postmarketing cases of hepatosplenic T-cell lymphoma, a rare type of T-cell lymphoma, have been reported in patients treated with TNF blockers, including REMICADE®. These cases have had a very aggressive disease course and have been fatal. The majority of reported REMICADE® cases have occurred in patients with Crohn’s disease or ulcerative colitis and most were in adolescent and young adult males. Almost all of these patients had received treatment with azathioprine or 6-mercaptopurine concomitantly with REMICADE® at or prior to diagnosis. Carefully assess the risks and benefits of treatment with REMICADE®, especially in these patient types.
In clinical trials of all TNF blockers, more cases of lymphoma were observed compared with controls and the expected rate in the general population. However, patients with Crohn’s disease, rheumatoid arthritis, or plaque psoriasis may be at higher risk for developing lymphoma. In clinical trials of some TNF blockers, including REMICADE®, more cases of other malignancies were observed compared with controls. The rate of these malignancies among patients treated with REMICADE® was similar to that expected in the general population whereas the rate in control patients was lower than expected. Cases of acute and chronic leukemia have been reported with postmarketing TNF-blocker use. As the potential role of TNF blockers in the development of malignancies is not known, caution should be exercised when considering treatment of patients with a current or a past history of malignancy or other risk factors such as chronic obstructive pulmonary disease (COPD).
Melanoma and Merkel cell carcinoma have been reported in patients treated with TNF‑blocker therapy, including REMICADE®. Periodic skin examination is recommended for all patients, particularly those with risk factors for skin cancer.
A population-based retrospective cohort study found a 2- to 3-fold increase in the incidence of invasive cervical cancer in women with rheumatoid arthritis treated with REMICADE® compared to biologics-naïve patients or the general population, particularly those over 60 years of age. A causal relationship between REMICADE® and cervical cancer cannot be excluded. Periodic screening should continue in women treated with REMICADE®.
CONTRAINDICATIONS
The use of REMICADE® at doses >5 mg/kg is contraindicated in patients with moderate or severe heart failure. REMICADE® is contraindicated in patients with a previous severe hypersensitivity reaction to infliximab or any of the inactive ingredients of REMICADE® or any murine proteins (severe hypersensitivity reactions have included anaphylaxis, hypotension, and serum sickness).
HEPATITIS B REACTIVATION
TNF blockers, including REMICADE®, have been associated with reactivation of hepatitis B virus (HBV) in patients who are chronic carriers. Some cases were fatal. Patients should be tested for HBV infection before initiating REMICADE®. For patients who test positive, consult a physician with expertise in the treatment of hepatitis B. Exercise caution when prescribing REMICADE® for patients identified as carriers of HBV and monitor closely for active HBV infection during and following termination of therapy with REMICADE®. Discontinue REMICADE® in patients who develop HBV reactivation and initiate antiviral therapy with appropriate supportive treatment. Exercise caution when considering resumption of REMICADE® and monitor patients closely.
HEPATOTOXICITY
Severe hepatic reactions, including acute liver failure, jaundice, hepatitis, and cholestasis have been reported in patients receiving REMICADE® postmarketing. Some cases were fatal or required liver transplant. Aminotransferase elevations were not noted prior to discovery of liver injury in many cases. Patients with symptoms or signs of liver dysfunction should be evaluated for evidence of liver injury. If jaundice and/or marked liver enzyme elevations (eg, ≥5 times the upper limit of normal) develop, REMICADE® should be discontinued, and a thorough investigation of the abnormality should be undertaken.
HEART FAILURE
In a randomized, placebo-controlled study in patients with moderate or severe heart failure (NYHA Functional Class III/IV), higher mortality rates and a higher risk of hospitalization were observed at Week 28 at a dose of 10 mg/kg and higher rates of cardiovascular events were observed at both 5 mg/kg and 10 mg/kg. There have been postmarketing reports of new onset and worsening heart failure, with and without identifiable precipitating factors. Patients with moderate or severe heart failure taking REMICADE® (≤5 mg/kg) or patients with mild heart failure should be closely monitored and treatment should be discontinued if new or worsening symptoms appear.
HEMATOLOGIC EVENTS
Cases of leukopenia, neutropenia, thrombocytopenia, and pancytopenia (some fatal) have been reported. The causal relationship to REMICADE® therapy remains unclear. Exercise caution in patients who have ongoing or a history of significant hematologic abnormalities. Advise patients to seek immediate medical attention if they develop signs and symptoms of blood dyscrasias or infection. Consider discontinuation of REMICADE® in patients who develop significant hematologic abnormalities.
HYPERSENSITIVITY
REMICADE® has been associated with hypersensitivity reactions that differ in their time of onset. Anaphylaxis, acute urticaria, dyspnea, and hypotension have occurred in association with infusions of REMICADE®. Medications for the treatment of hypersensitivity reactions should be available.
CARDIOVASCULAR AND CEREBROVASCULAR REACTIONS DURING AND AFTER INFUSION
Serious cerebrovascular accidents, myocardial ischemia/infarction (some fatal), hypotension, hypertension, and arrhythmias have been reported during and within 24 hours of initiation of REMICADE® infusion. Cases of transient visual loss have been reported during or within 2 hours of REMICADE® infusion. Monitor patients during infusion and if a serious reaction occurs, discontinue infusion. Manage reactions according to signs and symptoms.
NEUROLOGIC EVENTS
TNF blockers, including REMICADE®, have been associated with CNS manifestation of systemic vasculitis, seizure, and new onset or exacerbation of CNS demyelinating disorders, including multiple sclerosis and optic neuritis, and peripheral demyelinating disorders, including Guillain- Barré syndrome. Exercise caution when considering REMICADE® in patients with these disorders and consider discontinuation if these disorders develop.
CONCURRENT ADMINISTRATION WITH OTHER BIOLOGICS
Concurrent use of REMICADE® with anakinra, abatacept, tocilizumab, or other biologics used to treat the same conditions as REMICADE® is not recommended because of the possibility of an increased risk of infection. Care should be taken when switching from one biologic to another, since overlapping biological activity may further increase the risk of infection.
AUTOIMMUNITY
Treatment with REMICADE® may result in the formation of autoantibodies and in the development of a lupus-like syndrome. Discontinue treatment if symptoms of a lupus-like syndrome develop.
VACCINATIONS AND USE OF LIVE VACCINES/THERAPEUTIC INFECTIOUS AGENTS
Prior to initiating REMICADE®, update vaccinations in accordance with current vaccination guidelines. Live vaccines or therapeutic infectious agents should not be given with REMICADE® due to the possibility of clinical infections, including disseminated infections.
At least a 6-month waiting period following birth is recommended before the administration of any live vaccine to infants exposed in utero to REMICADE®.
ADVERSE REACTIONS
In clinical trials, the most common adverse reactions occurring in >10% of REMICADE®-treated patients included infections (eg, upper respiratory, sinusitis, and pharyngitis), infusion-related reactions, headache, and abdominal pain.
For more information, please see the full Prescribing Information and Medication Guide for REMICADE®. Provide the Medication Guide to your patients and encourage discussion.
IMPORTANT SAFETY INFORMATION
SERIOUS INFECTIONS
Patients treated with SIMPONI ARIA® (golimumab) are at increased risk for developing serious infections that may lead to hospitalization or death. Most patients who developed these infections were taking concomitant immunosuppressants such as methotrexate or corticosteroids. Discontinue SIMPONI ARIA® if a patient develops a serious infection.
Reported infections with TNF blockers, of which SIMPONI ARIA® is a member, include:
- Active tuberculosis (TB), including reactivation of latent TB. Patients frequently presented with disseminated or extrapulmonary disease. Test patients for latent TB before SIMPONI ARIA® use and during therapy. Initiate treatment for latent infection prior to SIMPONI ARIA® use.
- Invasive fungal infections, including histoplasmosis, coccidioidomycosis, candidiasis, aspergillosis, blastomycosis, and pneumocystosis. Patients with histoplasmosis or other invasive fungal infections may present with disseminated, rather than localized, disease. Consider empiric anti-fungal therapy in patients at risk for invasive fungal infections who develop severe systemic illness.
- Bacterial, viral, and other infections due to opportunistic pathogens, including Legionella and Listeria.
Consider the risks and benefits of treatment with SIMPONI ARIA® prior to initiating therapy in patients with chronic or recurrent infection. Do not start SIMPONI ARIA® in patients with clinically important active infections, including localized infections. Closely monitor patients for the development of signs and symptoms of infection during and after treatment with SIMPONI ARIA®, including the possible development of TB in patients who tested negative for latent TB infection prior to initiating therapy, who are on treatment for latent TB, or who were previously treated for TB infection.
Risk of infection may be higher in patients greater than 65 years of age, patients with co-morbid conditions and/or patients taking concomitant immunosuppressant therapy. Other serious infections observed in patients treated with SIMPONI ARIA® included sepsis, pneumonia, cellulitis, and abscess.
MALIGNANCIES
cp-51207v3Malignancies, some fatal, have been reported in children, adolescents, and young adult patients treated with golimumab. Approximately half the cases were lymphomas, including Hodgkin’s and non-Hodgkin’s lymphoma. The other cases represented a variety of malignancies, including rare malignancies usually associated with immunosuppression and malignancies not usually observed in children or adolescents. Malignancies occurred after a median of 30 months after the first dose of therapy. Most of the patients were receiving concomitant immunosuppressants.
In the controlled portions of clinical trials of TNF blockers including the subcutaneous formulation of golimumab, more cases of lymphoma have been observed among patients receiving anti-TNF treatment compared with patients in the control groups. In clinical trials, the incidence of malignancies other than lymphoma and non-melanoma skin cancer per 100 patient-years of follow-up was 0.56 (95% CI: 0.01, 3.11) in the SIMPONI ARIA® group compared with an incidence of 0 (95% CI: 0.00, 3.79) in the placebo group. Cases of acute and chronic leukemia have been reported with TNF-blocker use, including SIMPONI ARIA®. The risks and benefits of TNF-blocker therapy should be considered prior to initiating therapy in patients with a known malignancy or who develop a malignancy.
Postmarketing cases of hepatosplenic T-cell lymphoma (HSTCL), a rare type of T-cell lymphoma, have been reported in patients treated with TNF blockers. These cases have had a very aggressive disease course and have been fatal. Nearly all reported cases have occurred in patients with Crohn’s disease or ulcerative colitis, and the majority were in adolescent and young adult males. Almost all of these patients had received treatment with azathioprine or 6-mercaptopurine concomitantly with a TNF blocker at or prior to diagnosis. A risk for the development for HSTCL in patients treated with TNF blockers cannot be excluded.
Melanoma and Merkel cell carcinoma have been reported in patients treated with TNF-blocking agents, including SIMPONI ARIA®. Periodic skin examination is recommended for all patients, particularly those with risk factors for skin cancer.
HEPATITIS B REACTIVATION
The use of TNF blockers, of which SIMPONI ARIA® is a member, has been associated with reactivation of hepatitis B virus (HBV) in patients who are chronic hepatitis B carriers. In some instances, HBV reactivation occurring in conjunction with TNF-blocker therapy has been fatal. The majority of these reports have occurred in patients who received concomitant immunosuppressants.
All patients should be tested for HBV infection before initiating TNF-blocker therapy. For patients who test positive for hepatitis B surface antigen, consult a physician with expertise in the treatment of hepatitis B before initiating TNF-blocker therapy. Exercise caution when prescribing SIMPONI ARIA® for patients identified as carriers of HBV and closely monitor for active HBV infection during and following termination of therapy with SIMPONI ARIA®. Discontinue SIMPONI ARIA® in patients who develop HBV reactivation, and initiate antiviral therapy with appropriate supportive treatment. Exercise caution when considering resumption of SIMPONI ARIA®, and monitor patients closely.
CONGESTIVE HEART FAILURE
Cases of worsening congestive heart failure (CHF) and new-onset CHF have been reported with TNF blockers, including SIMPONI ARIA®. Some cases had a fatal outcome. Exercise caution in CHF patients receiving SIMPONI ARIA® and monitor them closely during therapy. Discontinue SIMPONI ARIA® if new or worsening symptoms of heart failure appear.
DEMYELINATING DISORDERS
Use of TNF blockers, including SIMPONI ARIA®, has been associated with rare cases of new-onset or exacerbation of demyelinating disorders, including multiple sclerosis (MS) and Guillain-Barré syndrome. Cases of central demyelination, MS, optic neuritis, and peripheral demyelinating polyneuropathy have rarely been reported in patients treated with golimumab. Exercise caution in considering the use of SIMPONI ARIA® in patients with these disorders. Consider discontinuation if these disorders develop.
AUTOIMMUNITY
Treatment with TNF blockers, including SIMPONI ARIA®, may result in the formation of antinuclear antibodies. Rarely, treatment with TNF blockers may result in a lupus-like syndrome. Discontinue treatment if symptoms of a lupus-like syndrome develop.
USE WITH OTHER DRUGS
The concomitant use of a TNF blocker and abatacept or anakinra was associated with a higher risk of serious infections, therefore the use of SIMPONI ARIA® in combination with these products is not recommended. Care should be taken when switching from one biologic to another since overlapping biological activity may further increase the risk of infection. A higher rate of serious infections has also been observed in RA patients treated with rituximab who received subsequent treatment with a TNF blocker. The concomitant use of SIMPONI ARIA® with biologics approved to treat RA is not recommended because of the possibility of an increased risk of infection.
HEMATOLOGIC CYTOPENIAS
There have been reports of pancytopenia, leukopenia, neutropenia, agranulocytosis, aplastic anemia, and thrombocytopenia in patients receiving SIMPONI ARIA®. Exercise caution when using SIMPONI ARIA® in patients who have or had significant cytopenias.
VACCINATIONS/THERAPEUTIC INFECTIOUS AGENTS
Live vaccines or therapeutic infectious agents should not be given with SIMPONI ARIA® due to the possibility of clinical infections, including disseminated infections.
Update vaccinations prior to initiation of treatment in accordance with current vaccination guidelines. Advise patients to discuss with the physician before seeking any immunizations. At least a 6-month waiting period following birth is recommended before the administration of any live vaccine to infants exposed in utero to SIMPONI ARIA®.
HYPERSENSITIVITY REACTIONS
Serious systemic hypersensitivity reactions (including anaphylaxis) have been reported following administration of the subcutaneous formulation of golimumab and SIMPONI ARIA®, some occurring after the first dose. Hypersensitivity reactions including hives, pruritus, dyspnea, and nausea, were reported in association with infusions of SIMPONI ARIA®. If an anaphylactic or other serious allergic reaction occurs, discontinue SIMPONI ARIA® immediately and institute appropriate therapy.
ADVERSE REACTIONS
The most serious adverse reactions were serious infections and malignancies.
The most common adverse reactions (incidence ≥3%) reported in clinical trials were: upper respiratory tract infection, alanine aminotransferase increase, viral infection, aspartate aminotransferase increase, neutrophil count decrease, bronchitis, hypertension, and rash. In the controlled phase of Trial RA, the rate of infusions associated with an infusion reaction was reported in 1.1% of SIMPONI ARIA® infusions compared with 0.2% of infusions in the control group.
The adverse reactions observed in pediatric patients with polyarticular Juvenile Idiopathic Arthritis (pJIA) were consistent with the established safety profile of SIMPONI ARIA® in adult patients with RA and PsA.
Please see the full Prescribing Information and Medication Guide for SIMPONI ARIA®. Provide the Medication Guide to your patients and encourage discussion.
Indication for
SIMPONI ARIA® (golimumab)
INDICATION
SIMPONI ARIA® is indicated for the treatment of adults with moderately to severely active rheumatoid arthritis (RA) in combination with methotrexate (MTX).
IMPORTANT SAFETY INFORMATION
SERIOUS INFECTIONS
Patients treated with REMICADE® (infliximab) are at increased risk for developing serious infections that may lead to hospitalization or death. Most patients who developed these infections were taking concomitant immunosuppressants such as methotrexate or corticosteroids. Discontinue REMICADE® if a patient develops a serious infection or sepsis.
Reported infections include:
- Active tuberculosis (TB), including reactivation of latent TB. Patients frequently presented with disseminated or extrapulmonary disease. Patients should be tested for latent TB before and during treatment with REMICADE®.1,2 Treatment for latent infection should be initiated prior to treatment with REMICADE®.
- Invasive fungal infections, including histoplasmosis, coccidioidomycosis, candidiasis, aspergillosis, blastomycosis, pneumocystosis, and cryptococcosis. Patients may present with disseminated, rather than localized, disease. Empiric anti-fungal therapy should be considered in patients at risk for invasive fungal infections who develop severe systemic illness.
- Bacterial, viral, and other infections due to opportunistic pathogens, including Legionella, Listeria, and Salmonella.
The risks and benefits of treatment with REMICADE® should be carefully considered prior to initiating therapy in patients with chronic or recurrent infection. Closely monitor patients for the development of signs and symptoms of infection during and after treatment with REMICADE®, including the possible development of TB in patients who tested negative for latent TB infection prior to initiating therapy, who are on treatment for latent TB, or who were previously treated for TB infection.
Risk of infection may be higher in patients greater than 65 years of age, pediatric patients, patients with co-morbid conditions and/or patients taking concomitant immunosuppressant therapy. In clinical trials, other serious infections observed in patients treated with REMICADE® included pneumonia, cellulitis, abscess, and skin ulceration.
MALIGNANCIES
cp-62063v2Lymphoma and other malignancies, some fatal, have been reported in children and adolescent patients treated with TNF blockers, including REMICADE®. Approximately half of these cases were lymphomas, including Hodgkin’s and non-Hodgkin’s lymphoma. The other cases represented a variety of malignancies, including rare malignancies that are usually associated with immunosuppression and malignancies that are not usually observed in children and adolescents. The malignancies occurred after a median of 30 months after the first dose of therapy. Most of the patients were receiving concomitant immunosuppressants.
Postmarketing cases of hepatosplenic T-cell lymphoma, a rare type of T-cell lymphoma, have been reported in patients treated with TNF blockers, including REMICADE®. These cases have had a very aggressive disease course and have been fatal. The majority of reported REMICADE® cases have occurred in patients with Crohn’s disease or ulcerative colitis and most were in adolescent and young adult males. Almost all of these patients had received treatment with azathioprine or 6-mercaptopurine concomitantly with REMICADE® at or prior to diagnosis. Carefully assess the risks and benefits of treatment with REMICADE®, especially in these patient types.
In clinical trials of all TNF blockers, more cases of lymphoma were observed compared with controls and the expected rate in the general population. However, patients with Crohn’s disease, rheumatoid arthritis, or plaque psoriasis may be at higher risk for developing lymphoma. In clinical trials of some TNF blockers, including REMICADE®, more cases of other malignancies were observed compared with controls. The rate of these malignancies among patients treated with REMICADE® was similar to that expected in the general population whereas the rate in control patients was lower than expected. Cases of acute and chronic leukemia have been reported with postmarketing TNF-blocker use. As the potential role of TNF blockers in the development of malignancies is not known, caution should be exercised when considering treatment of patients with a current or a past history of malignancy or other risk factors such as chronic obstructive pulmonary disease (COPD).
Melanoma and Merkel cell carcinoma have been reported in patients treated with TNF‑blocker therapy, including REMICADE®. Periodic skin examination is recommended for all patients, particularly those with risk factors for skin cancer.
A population-based retrospective cohort study found a 2- to 3-fold increase in the incidence of invasive cervical cancer in women with rheumatoid arthritis treated with REMICADE® compared to biologics-naïve patients or the general population, particularly those over 60 years of age. A causal relationship between REMICADE® and cervical cancer cannot be excluded. Periodic screening should continue in women treated with REMICADE®.
CONTRAINDICATIONS
The use of REMICADE® at doses >5 mg/kg is contraindicated in patients with moderate or severe heart failure. REMICADE® is contraindicated in patients with a previous severe hypersensitivity reaction to infliximab or any of the inactive ingredients of REMICADE® or any murine proteins (severe hypersensitivity reactions have included anaphylaxis, hypotension, and serum sickness).
HEPATITIS B REACTIVATION
TNF blockers, including REMICADE®, have been associated with reactivation of hepatitis B virus (HBV) in patients who are chronic carriers. Some cases were fatal. Patients should be tested for HBV infection before initiating REMICADE®. For patients who test positive, consult a physician with expertise in the treatment of hepatitis B. Exercise caution when prescribing REMICADE® for patients identified as carriers of HBV and monitor closely for active HBV infection during and following termination of therapy with REMICADE®. Discontinue REMICADE® in patients who develop HBV reactivation and initiate antiviral therapy with appropriate supportive treatment. Exercise caution when considering resumption of REMICADE® and monitor patients closely.
HEPATOTOXICITY
Severe hepatic reactions, including acute liver failure, jaundice, hepatitis, and cholestasis have been reported in patients receiving REMICADE® postmarketing. Some cases were fatal or required liver transplant. Aminotransferase elevations were not noted prior to discovery of liver injury in many cases. Patients with symptoms or signs of liver dysfunction should be evaluated for evidence of liver injury. If jaundice and/or marked liver enzyme elevations (eg, ≥5 times the upper limit of normal) develop, REMICADE® should be discontinued, and a thorough investigation of the abnormality should be undertaken.
HEART FAILURE
In a randomized, placebo-controlled study in patients with moderate or severe heart failure (NYHA Functional Class III/IV), higher mortality rates and a higher risk of hospitalization were observed at Week 28 at a dose of 10 mg/kg and higher rates of cardiovascular events were observed at both 5 mg/kg and 10 mg/kg. There have been postmarketing reports of new onset and worsening heart failure, with and without identifiable precipitating factors. Patients with moderate or severe heart failure taking REMICADE® (≤5 mg/kg) or patients with mild heart failure should be closely monitored and treatment should be discontinued if new or worsening symptoms appear.
HEMATOLOGIC EVENTS
Cases of leukopenia, neutropenia, thrombocytopenia, and pancytopenia (some fatal) have been reported. The causal relationship to REMICADE® therapy remains unclear. Exercise caution in patients who have ongoing or a history of significant hematologic abnormalities. Advise patients to seek immediate medical attention if they develop signs and symptoms of blood dyscrasias or infection. Consider discontinuation of REMICADE® in patients who develop significant hematologic abnormalities.
HYPERSENSITIVITY
REMICADE® has been associated with hypersensitivity reactions that differ in their time of onset. Anaphylaxis, acute urticaria, dyspnea, and hypotension have occurred in association with infusions of REMICADE®. Medications for the treatment of hypersensitivity reactions should be available.
CARDIOVASCULAR AND CEREBROVASCULAR REACTIONS DURING AND AFTER INFUSION
Serious cerebrovascular accidents, myocardial ischemia/infarction (some fatal), hypotension, hypertension, and arrhythmias have been reported during and within 24 hours of initiation of REMICADE® infusion. Cases of transient visual loss have been reported during or within 2 hours of REMICADE® infusion. Monitor patients during infusion and if a serious reaction occurs, discontinue infusion. Manage reactions according to signs and symptoms.
NEUROLOGIC EVENTS
TNF blockers, including REMICADE®, have been associated with CNS manifestation of systemic vasculitis, seizure, and new onset or exacerbation of CNS demyelinating disorders, including multiple sclerosis and optic neuritis, and peripheral demyelinating disorders, including Guillain- Barré syndrome. Exercise caution when considering REMICADE® in patients with these disorders and consider discontinuation if these disorders develop.
CONCURRENT ADMINISTRATION WITH OTHER BIOLOGICS
Concurrent use of REMICADE® with anakinra, abatacept, tocilizumab, or other biologics used to treat the same conditions as REMICADE® is not recommended because of the possibility of an increased risk of infection. Care should be taken when switching from one biologic to another, since overlapping biological activity may further increase the risk of infection.
AUTOIMMUNITY
Treatment with REMICADE® may result in the formation of autoantibodies and in the development of a lupus-like syndrome. Discontinue treatment if symptoms of a lupus-like syndrome develop.
VACCINATIONS AND USE OF LIVE VACCINES/THERAPEUTIC INFECTIOUS AGENTS
Prior to initiating REMICADE®, update vaccinations in accordance with current vaccination guidelines. Live vaccines or therapeutic infectious agents should not be given with REMICADE® due to the possibility of clinical infections, including disseminated infections.
At least a 6-month waiting period following birth is recommended before the administration of any live vaccine to infants exposed in utero to REMICADE®.
ADVERSE REACTIONS
In clinical trials, the most common adverse reactions occurring in >10% of REMICADE®-treated patients included infections (eg, upper respiratory, sinusitis, and pharyngitis), infusion-related reactions, headache, and abdominal pain.
For more information, please see the full Prescribing Information and Medication Guide for REMICADE®. Provide the Medication Guide to your patients and encourage discussion.
Indication for
REMICADE® (infliximab)
INDICATION
REMICADE®, in combination with methotrexate, is indicated for reducing signs and symptoms, inhibiting the progression of structural damage, and improving physical function in adult patients with moderately to severely active rheumatoid arthritis (RA).